3-Chloroperoxybenzoic acid, m-CPBA

Product Manager:Nick Wilde
m-CPBA, a potent oxidizing agent, rivals other peracids in its strength. One notable advantage of 3-chloroperbenzoic acid lies in its handling convenience, being available in powdered form that can be safely stored in refrigerated conditions. However, commercially, achieving a purity level exceeding 75% is rarely feasible due to the inherent instability of the pure compound. Consequently, transporting MCPBA with a concentration above 72% by air is prohibited. In terms of environmental concerns, the primary contaminant of MCPBA is 3-chlorobenzoic acid, which may account for up to 10% of the mixture, while water is also added for safety reasons.

MCPBA, a versatile peracid, finds widespread application in laboratory settings. Its primary utility lies in oxidizing various chemical compounds, including:
• Aldehydes and ketones, which are converted to esters through the Bayer-Villiger Oxidation
• Olefins, which are transformed into epoxides
• Sulfides, undergoing oxidation to yield sulfoxides and sulfones
• Amines, undergoing transformations to nitroalkanes, nitroxides, or N-oxides
Despite its exceptional reactivity and selectivity in many reactions, the use of MCPBA in industrial production is discouraged due to considerations of atomic economy. Research in this field is increasingly focused on exploring the use of hydrogen peroxide, often in conjunction with tailored catalysts or in situ generated, simpler peracids like peracetic acid or potassium peroxymonosulfate. Nonetheless, in numerous reactions, MCPBA exhibits remarkable selectivity that surpasses that of hydrogen peroxide and other peracids.
Name Reactions

Cope Elimination
Prilezhaev Reaction

Rubottom Oxidation
Recent Literature

Employing a solvent with a higher density than the fluorous phase presents an alternative strategy to the U-tube method in phase-vanishing reactions, particularly when both reactants are less dense than the fluorous phase. This approach allows for effective mixing and reaction of the components without relying on the traditional U-tube setup.
N. K. Jana, J. G. Verkade, Org. Lett., 2003, 5, 3787-3790.
https://doi.org/10.1021/ol035391b

N. K. Jana, J. G. Verkade, Org. Lett., 2003, 5, 3787-3790.
https://doi.org/10.1021/ol035391b

N. K. Jana, J. G. Verkade, Org. Lett., 2003, 5, 3787-3790.
https://doi.org/10.1021/ol035391b

We present the outcomes of a highly diastereoselective epoxidation process, focusing on allylic diols derived from Baylis-Hillman adducts.
R. S. Porto, M. L. A. A. Vasconcellos, E. Ventura, F. Coelho, Synthesis, 2005, 2297-2306.
https://doi.org/10.1055/s-2005-872091

A metal-free and environmentally friendly approach to diacetoxylation of alkenes, employing readily available peroxyacids as oxidizing agents, has been developed. Catalyzed by triflic acid, this method offers a clean and efficient route, further enabling oxidative lactonization of unsaturated carboxylic acids with good to high yields.
Y.-B. Kang, L. H. Gade, J. Org. Chem., 2012, 77, 1610-1615.
https://doi.org/10.1021/jo202491y

A streamlined, one-pot process for the stereospecific conversion of 1,2-disubstituted olefins into their corresponding disubstituted cyclic carbonates involves sequential treatment with m-CPBA, CBr3CO2H, and DBU. Similarly, when a secondary allylic or homoallylic amine is initially reacted with CBr3CO2H in solution, followed by sequential addition of m-CPBA and DBU, the resulting product is a cyclic carbamate. This methodology offers a concise and efficient route to these valuable cyclic derivatives.
S. G. Davies, A. M. Fletcher, W. Kurosawa, J. A. Lee, G. Poce, P. M. Roberts, J. E. Thomson, D. M. Williamson, J. Org. Chem., 2010, 75, 7745-7756.
https://doi.org/10.1021/jo101614f

An efficient method for the synthesis of amides in high yields involves the oxidation of aldimines, which can be readily derived from aldehydes. This process utilizes m-CPBA and BF3·OEt2 as catalysts and produces amides in a manner that is strongly influenced by the electron-releasing properties of the aromatic substituent (Ar) present in the imine precursor.
G. An, M. Kim, J. Y. Kim, H. Rhee, Tetrahedron Lett., 2003, 44, 2183-2186.
https://doi.org/10.1016/S0040-4039(03)00156-4

The selective formation of cyclopropylketones can be achieved through an oxidative ring contraction of readily accessible cyclobutene derivatives, utilizing mCPBA as the oxidant. This functional group-tolerant transformation occurs under mild conditions at room temperature.
A. N. Baumann, F. Schüppel, M. Eisold, A. Kreppel, R. de Vivie-Riedle, D. Didier, J. Org. Chem., 2018, 83, 4905-4921.
https://doi.org/10.1021/acs.joc.8b00297

Gold-catalyzed tandem cycloisomerization/oxidation of homopropargyl alcohols offers a straightforward route to various γ-lactones. This methodology differs significantly from related ruthenium-catalyzed reactions, which proceed via a ruthenium vinylidene intermediate, underscoring the unique mechanism of the gold-catalyzed process.
C. Shu, M.-Q. Liu, Y-Z. Sun, L.-W. Ye, Org. Lett., 2012, 14, 4958-4961.
https://doi.org/10.1021/ol302323a

A streamlined one-pot γ-lactonization of homopropargyl alcohols is achieved through an alkyne deprotonation/boronation/oxidation sequence. In this process, oxidation of the resulting alkynyl boronate generates a ketene intermediate, which is subsequently trapped by the adjacent hydroxy group, leading to the formation of the desired γ-lactone.
D. Yamane, H. Tanaka, A. Hirata, Y. Tamura, D. Takahashi, Y. Takahashi, T. Nagamitsu, M. Ohtawa, Org. Lett., 2021, 23, 2831-2835.
https://doi.org/10.1021/acs.orglett.1c00840

The Baeyer-Villiger oxidation can be harnessed for enantioselective desymmetrization of meso cyclic ketones, as well as for the kinetic resolution of racemic 2-arylcyclohexanones. This methodology allows for the enantioselective transformation of these compounds into their corresponding ester derivatives with high selectivity and efficiency.
L. Zhou, X. H. Liu, J. Ji, Y. H. Zhang, X. L. Hu, L. L. Lin, X. M. Feng, J. Am. Chem. Soc., 2012, 134, 17023-17026.
https://doi.org/10.1021/ja309262f

The efficiency of iodoarene amide catalysts in the α-oxytosylation of propiophenone is governed by both steric and electronic factors. By utilizing a highly reactive meta-substituted benzamide catalyst, a range of substituted propiophenones can be transformed into α-tosyloxy ketones in excellent isolated yields.
T. R. Lex, M. I. Swasy, D. C. Whitehead, J. Org. Chem., 2015, 80, 12234-12243.
https://doi.org/10.1021/acs.joc.5b02129

A versatile method for the synthesis of α-tosyloxy ketones involves reacting various ketones with a mixture containing MCPBA, PTSA•H2O, catalytic amounts of iodine, tert-butylbenzene, and a solvent blend of acetonitrile and 2,2,2-trifluoroethanol. This process initially generates 4-tert-butyl-1-iodobenzene, which is then converted into the α-tosyloxylation reagent 4-tert-butyl-1-[(hydroxy)(tosyloxy)iodo]benzene through a reaction with MCPBA and PTSA•H2O.
A. Tanaka, K. Moriyama, H. Togo, Synlett, 2011, 1853-1854.
https://doi.org/10.1055/s-0030-1260948

High yields of various α-tosyloxyketones were achieved through the reaction of ketones with m-chloroperbenzoic acid and p-toluenesulfonic acid, facilitated by a catalytic amount of iodobenzene. This method offers an efficient route to the synthesis of α-tosyloxyketones.
Y. Yamamoto, H. Togo, Synlett, 2006, 798-800.
https://doi.org/10.1055/s-2006-933120

An environmentally benign and metal-free method for the oxidative rearrangement of chalcones utilizes in situ generated hypoiodite as the catalyst. This process operates under mild conditions and avoids the need for rare or toxic metals, making it a favorable approach for organic synthesis.
M. Zheng, C. Huang, J.-Z. Yan, S.-L. Xie, S.-J. Ke, H.-D. Xia, Y.-N. Duan, J. Org. Chem., 2023, 88, 1504-1514.
https://doi.org/10.1021/acs.joc.2c02291

A versatile Ag2O-catalyzed reaction allows for the synthesis of α-carbonyloxy esters from carboxylic acids, ynol ethers, and m-CPBA. This protocol features the formation of three C-O bonds and utilizes readily available starting materials, enabling a broad substrate scope for the synthesis of these valuable esters.
L. Zeng, H. Sajiki, S. Cui, Org. Lett., 2019, 21, 6423-6426.
https://doi.org/10.1021/acs.orglett.9b02323

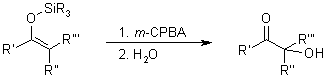
A straightforward metal-free method for the α-hydroxylation of α-unsubstituted β-oxoesters and β-oxoamides utilizes m-chloroperbenzoic acid as the oxidant. This approach provides access to important α-hydroxy-β-dicarbonyl moieties under mild reaction conditions and avoids the use of metals. Additionally, the hydroxylated products can be easily transformed into vicinal tricarbonyl compounds, which are valuable intermediates for further synthesis.
H. Asahara, N. Nishiwaki, J. Org. Chem., 2014, 79, 11735-11739.
https://doi.org/10.1021/jo501985u

A highly efficient method for the synthesis of primary, secondary, and tertiary alkane-, arene-, and heteroarenesulfonamides involves the reaction of methyl sulfinates with lithium amides, followed by oxidation of the resulting sulfinamides. This protocol is advantageous as it avoids the use of hazardous, unstable, or volatile reagents and maintains the configurational stability of the amines, leading to high yields of the desired sulfonamide products.
J. L. C. Ruano, A. Parra, F. Yuste, V. M. Mastranzo, Synthesis, 2008, 311-312.
https://doi.org/10.1055/s-2007-1000850

The oxidation of β-piperidinoethylsulfides with m-chloroperbenzoic acid generates intermediates featuring both N-oxide and sulfone functionalities. These intermediates undergo a Cope-type elimination to yield vinylsulfones, which can then be captured by amines to produce β-aminoethylsulfones. This synthetic methodology is adaptable to multiple-parallel formats and holds promise for a wide range of applications in medicinal chemistry.
R. J. Gruffin, A. Henderson, N. J. Curtin, A. Echalier, J. A. Endicott, I. R. Hardcastle, D. R. Newell, M. E. M. Noble, L.-Z. Wang, B. T. Golding, J. Am. Chem. Soc., 2006, 128, 6012-6013.
https://doi.org/10.1021/ja060595j

The synthesis of N-cyanosulfilimines is efficiently accomplished through the reaction of sulfides with cyanogen amine in the presence of a base and a halogenating agent such as NBS or I2. Subsequent oxidation and decyanation of the resulting intermediates leads to the formation of synthetically valuable sulfoximines.
O. García Mancheño, O. Bistri, C. Bolm, Org. Lett., 2007, 9, 3809-3811.
https://doi.org/10.1021/ol7016577

Iodobenzene serves as a recyclable catalyst in conjunction with m-chloroperbenzoic acid as the terminal oxidant, enabling an efficient and regioselective monobromination of electron-rich aromatic compounds. This process, which utilizes lithium bromide as the brominating agent, proceeds rapidly in tetrahydrofuran at room temperature, yielding regioselective monobrominated products in good yields.
Z. Zhou, X. He, Synthesis, 2011, 207-209.

The utilization of MCPBA results in a gentle yet highly productive method for synthesizing phenols from arylboronic acids in an aqueous medium at ambient temperatures. Studies involving isotopic labeling indicate that the hydroxyl oxygen atom present in the phenol product could potentially stem from the MCPBA.
D.-S. Chen, J.-M. Huang, Synlett, 2013, 24, 499-501.
https://doi.org/10.1055/s-0032-1318197

A versatile approach for the 1,3-oxidation of cyclopropanes, facilitated by aryl iodine(I-III) catalysis, offers a powerful platform for synthesizing diverse functional groups including 1,3-difluorides, fluoroacetoxylated derivatives, 1,3-diols, amino alcohols, and diamines. This strategy employs readily accessible and commercially available reagents, enabling its application to a broad range of substituted cyclopropane substrates, thereby expanding the synthetic toolbox for accessing these valuable compounds.
S. M. Banik, K. M. Mennie, E. N. Jacobsen, J. Am. Chem. Soc., 2017, 139, 9152-9155.
https://doi.org/10.1021/jacs.7b05160

Iodobenzene acts as an effective catalyst in promoting an oxidative cyclization reaction between Michael adducts of activated methylene compounds and nitroolefins or chalcones. This transformation, facilitated by the use of mCPBA as the terminal oxidant in conjunction with Bu4NI, yields a diverse array of highly functionalized cyclopropanes with remarkable diastereoselectivities.
Y. Li, H. Guo, R. Fan, Synthesis, 2020, 52, 928-932.
https://doi.org/10.1055/s-0039-1690809

A new, regiospecific, sequential one-pot synthesis of symmetrical and unsymmetrical diaryliodonium tetrafluoroborates, which are the most popular salts in metal-catalyzed arylations, is fast and high-yielding and has a large substrate scope. Furthermore, the corresponding diaryliodonium triflates can conveniently be obtained via an in situ anion exchange.
M. Bielawski, D. Aili, B. Olofsson, J. Org. Chem., 2008, 73, 4602-4607.
https://doi.org/10.1021/jo8004974

A direct and convenient route for synthesizing aryl(2,4,6-trimethoxyphenyl)iodonium trifluoroacetate salts from aryl iodides has been devised. This process utilizes stoichiometric amounts of trifluoroacetic acid as the counteranion and trimethoxybenzene as an auxiliary precursor, eliminating the need for a separate anion exchange step to incorporate the trifluoroacetate group. The reaction proceeds smoothly at mild temperatures and exhibits a wide substrate scope.
V. Carreras, A. H. Sandtorv, D. R. Stuart, J. Org. Chem., 2017, 82, 1279-1284.
https://doi.org/10.1021/acs.joc.6b02811

A rapid and efficient one-pot synthesis of aryl(2,4,6-trimethoxyphenyl)iodonium salts from aryl iodides, utilizing m-CPBA, p-toluenesulfonic acid, and trimethoxybenzene as reagents, has been developed. This method offers high yields and a wide substrate scope, showcasing its versatility. Furthermore, the utility of these synthesized salts is exemplified in arylation reactions involving C-, N-, O-, and S-nucleophiles, emphasizing their potential for use in diverse synthetic transformations.
T. L. Seidl, S. K. Sundalam, B. McCullough, D. R. Stuart, J. Org. Chem., 2016, 81, 1998-2009.
https://doi.org/10.1021/acs.joc.5b02833

A streamlined method for the efficient preparation of [(diacetoxy)iodo]arenes has been achieved through the treatment of iodoarenes with m-chloroperoxybenzoic acid in acetic acid. A notable advantage of this approach lies in its ability to facilitate the facile synthesis and isolation of [(diacetoxy)iodo]arenes adorned with electron-withdrawing groups, underscoring its practicality and versatility.
M. Iinuma, K. Moriyama, H. Togo, Synlett, 2012, 23, 2663-2666.
https://doi.org/10.1055/s-0032-1317345

One-pot syntheses of neutral and electron-rich [hydroxy(tosyloxy)iodo]arenes (HTIBs) from iodine and arenes avoid the need for expensive iodine(III) precursors. A large set of HTIBs, including a polyfluorinated analogue, can be obtained from the corresponding aryl iodides under mild conditions, without excess reagents, in high yields.
E. A. Merritt, V. M. T. Carneiro, L. F. Silva Jr., B. Olofsson, J. Org. Chem., 2010, 75, 7416-7419.
https://doi.org/10.1021/jo101227j

A streamlined direct synthesis of both symmetric and unsymmetric electron-dense diaryliodonium salts efficiently produces diaryliodonium tosylates in excellent yields, leveraging the combination of meta-chloroperoxybenzoic acid (MCPBA) and toluenesulfonic acid. Furthermore, an innovative in situ anion exchange process has been devised, allowing for the direct conversion of these tosylates into their corresponding triflate salts.
M. Zhu, N. Jalalian, B. Olofsson, Synlett, 2008, 592-596.
https://doi.org/10.1055/s-2008-1032050

This innovative gold-catalyzed tandem cycloisomerization/oxidation process, involving homopropargyl amides, offers a straightforward route to valuable chiral γ-lactams with exceptional enantiomeric excesses. By seamlessly integrating chiral tert-butylsulfinimine chemistry with gold catalysis, this method harnesses readily accessible starting materials, adheres to a simplified protocol, and operates under gentle reaction conditions, underscoring its practical appeal for synthetic applications.
C. Shu, M.-Q. Liu, S.-S. Wang, L. Li, L.-W. Ye, J. Org. Chem., 2013, 78, 3292-3299.
https://doi.org/10.1021/jo400127x

The utilization of m-chloroperoxybenzoic acid (m-CPBA) as an oxidant in ethyl acetate facilitates a swift and efficient oxidation of diverse aliphatic amines into oximes, achieving high conversion rates and exceeding 90% oxime selectivity. This transformation occurs without the need for a catalyst, operates at ambient temperature, and underscores the practicality of the method.
V. V. Patil, E. M. Gayakwad, G. S. Shankarling, J. Org. Chem., 2016, 81, 781-786.
https://doi.org/10.1021/acs.joc.5b01740

A streamlined one-pot, three-step methodology for crafting isoxazolines from aldehydes is presented. The protocol initiates with the conversion of aldehydes into aldoximes using hydroxylamine sulfate. Subsequently, these intermediates are oxidized to nitrile oxides by an on-the-spot generated hypervalent iodine species. Lastly, a 1,3-dipolar cycloaddition reaction between the nitrile oxides and alkenes efficiently yields isoxazolines in satisfactory quantities.
L. Han, B. Zhang, C. Xiang, J. Yan, Synthesis, 2014, 46, 503-509.
https://doi.org/10.1055/s-0033-1340464

A sequential transformation involving the oxidation of propargylamines to their corresponding oximes, followed by an intramolecular cyclization catalyzed by CuCl, enables the synthesis of a diverse array of isoxazoles. This method boasts broad functional group tolerance, making it a versatile tool for the construction of these heterocyclic scaffolds.
M. Duan, G. Hou, Y. Zhao, C. Zhu, C. Song, J. Org. Chem., 2022, 87, 11222-11225.
https://doi.org/10.1021/acs.joc.2c00896

An efficient iodine(III)-mediated oxidative cyclization of 2-hydroxystilbenes is achieved using 10 mol% of (diacetoxyiodo)benzene [PhI(OAc)2] as the catalyst, in conjunction with m-chloroperbenzoic acid. This protocol yields 2-arylbenzofurans in impressive to outstanding quantities, underscoring its effectiveness in constructing this valuable heterocyclic class.
F. V. Singh, S. R. Mangaonkar, Synthesis, 2018, 50, 4940-4948.
https://doi.org/10.1055/s-0037-1610650

Iodobenzene serves as an effective catalyst in the oxidative C-H amination of N″-aryl-N′-tosyl/N′-methylsulfonylamidines and N,N′-bis(aryl)amidines, utilizing m-chloroperoxybenzoic acid (mCPBA) as the terminal oxidant. This method, conducted at ambient temperature, is versatile and yields 1,2-disubstituted benzimidazoles in good quantities, highlighting its potential for the synthesis of these valuable compounds.
S. K. Alla, R. K. Kumar, P. Sadhu, T. Punniyamurthy, Org. Lett., 2013, 15, 1334-1337.
https://doi.org/10.1021/ol400274f

An organocatalyzed oxidative intramolecular C-N bond formation process involving phenylpropanamide derivatives results in the synthesis of 3,3-disubstituted oxindole derivatives in excellent yields. This highly productive reaction operates under transition metal-free and mild conditions, and its scalability to gram-scale demonstrates its practical applicability for the production of these valuable oxindole scaffolds.
Y. Wang, M. Yang, Y.-Y. Sun, Z.-G. Wu, H. Dai, S. Li, Org. Lett., 2021, 23, 8750-8754.
https://doi.org/10.1021/acs.orglett.1c03224

An enamine-catalyzed [3+2]-cycloaddition strategy involving aldehydes and N-hydroximidoyl chlorides, facilitated by triethylamine, yields 3,4,5-trisubstituted 5-(pyrrolidinyl)-4,5-dihydroisoxazoles. Subsequent oxidation of these cycloadducts provides a metal-free, high-yielding, and regiospecific pathway for the synthesis of 3,4-disubstituted isoxazoles, showcasing the versatility and efficiency of this approach.
Q.-f. Jia, P. M. S. Benjamin, J. Huang, Z. Du, X. Zheng, K. Zhang, A. H. Conney, J. Wang, Synlett, 2013, 24, 79-84.
https://doi.org/10.1055/s-0032-1317923
Quoted
from:https://www.organic-chemistry.org/chemicals/oxidations/meta-chloroperbenzoicacid.shtm
For more product details, please visit Aladdin Scientific website.