CRISPR/Cas9 and targeted genome editing
It is a long-term goal for biomedical researchers to develop efficient and reliable methods to make accurate and targeted changes in the genome of living cells. In recent years, a new tool based on bacterial CRISPR-associated protein-9 nuclease (Cas9) from Streptococcus pyogenes has aroused great interest among researchers1. This is after many attempts to manipulate gene function over the years, including homologous recombination2and RNA interference (RNAi)3. In particular, RNAi has become the main product in the laboratory, which can study gene function4,5 cheaply and with high throughput, but it is hindered by providing only temporary inhibition of gene function and unpredictable off-target effect6. Other recent methods of targeting genomic modification-zinc finger nuclease [ZFNs,7] and transcriptional activators such as effector nuclease [TALENs8]-enable researchers to produce permanent mutations by introducing double-strand breaks to activate repair pathways. The engineering cost of these methods is high and time-consuming, which limits their wide use, especially for large-scale and high-throughput research.
What is CRISPR/Cas9?
The functions of CRISPR (clusters of regularly spaced short palindromic repeat sequences) and CRISPR-related (Cas) genes are crucial in the adaptive immunity of selected bacteria and archaea, enabling organisms to respond to and eliminate invasive genetic material. These repeats were first found in E. coli9 in the 1980s, but it was not until 2007 that Barrangou and his colleagues confirmed their function, demonstrating that Streptococcus thermophilus can acquire phage resistance by integrating genomic fragments of infectious viruses into its CRISPR loci10.
Three types of CRISPR mechanisms have been identified, among which type II is the most studied. In this case, the invading DNA from the virus or plasmid is cut into small fragments and incorporated into the CRISPR locus in a series of short repeats (around 20 bps). The locus is transcribed, and then the transcript is processed to produce small RNA (crRNA-CRISPR RNA), which is used to guide the effect of endonuclease, targeting the invading DNA according to sequence complementarity (Figure 1)11.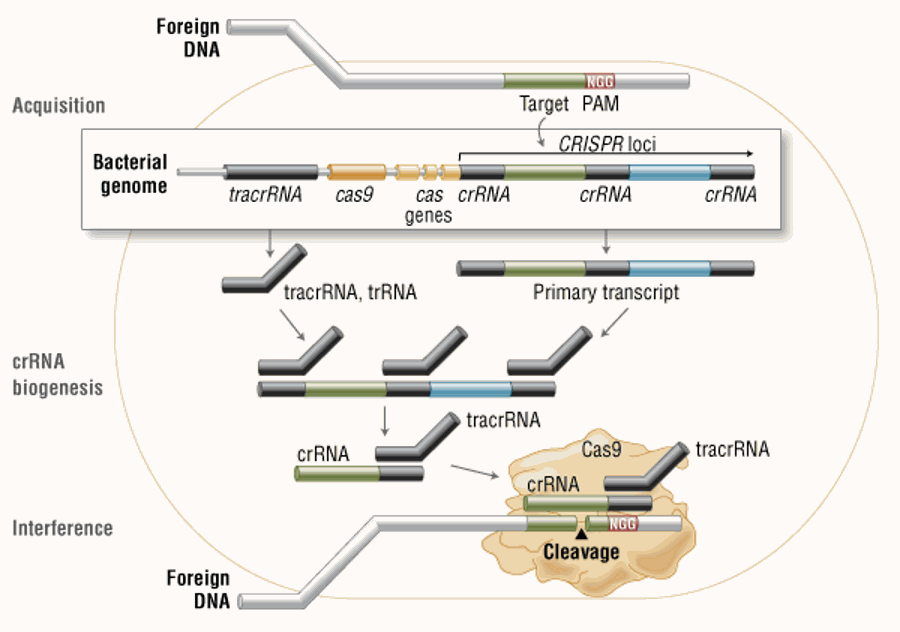
Figure 1. Cas9 in vivo: Bacterial Adaptive Immunity
Figure 1:[during the acquisition phase, the exogenous DNA is integrated into the bacterial genome at the CRISPR site. It is then transcribed and processed into crRNA in the process of crRNA biogenesis. In the process of interference, Cas9 endonuclease recombines with crRNA, and the isolated tracrRNA cleaves the foreign DNA containing 20 nucleotide crRNA complementary sequences adjacent to the PAM sequence.]
A Cas protein Cas9 (also known as Csn1) has been shown to be a key participant in some CRISPR mechanisms, especially the type II CRISPR system. Compared with other CRISPR systems, the type II CRISPR mechanism is unique because gene silencing requires only one Cas protein (Cas9)12. In type II system, Cas9 participates in the processing of crRNA12 and is responsible for the destruction of target DNA11. The function of Cas9 in these two steps depends on the existence of two nuclease domains, a RuvC-like nuclease domain located at the amino terminal and a HNH-like nuclease domain located in the middle region of the protein13.
In order to achieve site-specific DNA recognition and cleavage, Cas9 must be recombined with crRNA and a single transactivated crRNA (tracrRNA or trRNA), which is partially complementary to crRNA11. TracrRNA is necessary for crRNA maturation from primary transcripts that encode multiple pre-crRNA.This occurs in the presence of RNaseIII and Cas912. During the destruction of target DNA, two DNA strands were cleaved by HNH and RuvC-like ribonuclease domains, resulting in double strand breaks (DSB) at sites defined by 20 nucleotide target sequences in related crRNA transcripts. The HNH domain cuts complementary chains, while the RuvC domain cuts non-complementary chains. The double-stranded endonuclease activity of Cas9 also requires a short conserved sequence (2-5nts) called primordial spacer-related motif (PAM) to follow the 3'- of crRNA complementary sequence15. In fact, even completely complementary sequences are ignored by Cas9-RNA in the absence of PAM sequences.
Cas9 and CRISPR as New tools of Molecular Biology
The simplicity of type II CRISPR nuclease has only three essential components (Cas9 and crRNA and trRNA), making the system suitable for genome editing. This potential was realized by Doudna and Charpentier labs in 201211. Based on the type II CRISPR system described above, the author develops a simplified two-component system by combining trRNA and crRNA into a single synthetic single wizard RNA (sgRNA). Cas9 programmed by sgRNA is as effective in guiding targeted genetic changes as Cas9 programmed with separate trRNA and crRNA (Figure 2-A).
So far, three different variants of Cas9 nuclease have been used in genome editing programs. The first is wild-type Cas9, which can specifically cleave double-strand DNA at a fixed point, resulting in the activation of double-strand break (DSB) repair mechanism. DSB can repair17 through the cell non-homologous end junction (NHEJ) pathway, resulting in insertion and/or deletion (insertion deletion), thus destroying the target site. Alternatively, if a donor template is provided that is homologous to the target site, the DSB can be repaired through the homologous directed repair (HDR) pathway, allowing for precise substitution mutations (Figure 2-A)17,18.
Cong and his colleagues1further improved the accuracy of the Cas9 system by developing a mutant form, called Cas9D10A, which only has cleavage enzyme activity. This means that it cuts only one DNA chain and does not activate NHEJ. In contrast, when homologous repair templates are provided, DNA repair is carried out only through the high-fidelity HDR pathway, resulting in a reduction of insertion and deletion mutations1,11,19. Cas9D10A is more attractive in terms of target specificity when the site is designed to produce paired Cas9 complex targeting of adjacent DNA incisions20(see further details about “paired nickases” in Figure 2-B).
The third variant is nuclease deficient Cas9 (dCas9, Figure 2-C)21. Mutant H840A inactivated cleavage activity in HNH domain and D10A in RuvC domain, but did not prevent DNA binding11,21. Therefore, the variant can be used to specifically target any region of the genome without cutting. On the contrary, by fusing with various effect domains, dCas9 can be used as a tool for gene silencing or activation. In addition, it can be used as a visualization tool21,23-26. For example, Chen and his colleagues used dCas9 fused with enhanced green fluorescent protein (EGFP) to visualize repetitive DNA sequences with a single sgRNA or non-repetitive sites using multiple sgRNA27.
Figure 2. CRISPR/Cas9 System Applications
[Figure 2-A:wild-type Cas9 nuclease site-specific cleavage of double-stranded DNA activates double-strand break repair mechanism. In the absence of homologous repair template, non-homologous terminal connection will cause insertion deletion to destroy the target sequence. Alternatively, precise mutation and typing can be done by providing homologous repair templates and using homologous directed repair pathways.
Figure 2-B:mutant Cas9 producing site-specific single strand gap. Two sgRNA can be used to introduce staggered double strand breaks, and then homologous directed repair can be performed.
Figure 2-C:Cas9 lacking nuclease can be fused with various effect domains to achieve specific localization. For example, transcriptional activators, repressors and fluorescent proteins.]
Targeting efficiency and miss mutation
Targeting efficiency, or the percentage of expected mutations achieved, is one of the most important parameters for evaluating genome editing tools. The targeting efficiency of Cas9 is better than more mature methods, such as TALENs or ZFNs8. For example, in human cells, custom-designed ZFNs and TALENs can only achieve 1 to 50 percent efficiency29-31. In contrast, it is reported that the Cas9 system is more than 70% efficient in zebrafish32and plants33and 2-5% in inducing pluripotent stem cells34. In addition, Zhou and his colleagues were able to increase genome targeting to 78% in single-cell mouse embryos and achieve effective germline transmission by using double sgRNAs to target a single gene at the same time35.A widely used method for identifying mutations is the T7 endonuclease I mutation detection36,37(Figure 3). This detection method detects heteroduplex DNA, which is the result of annealing of required mutated DNA strands and wild-type DNA strands37.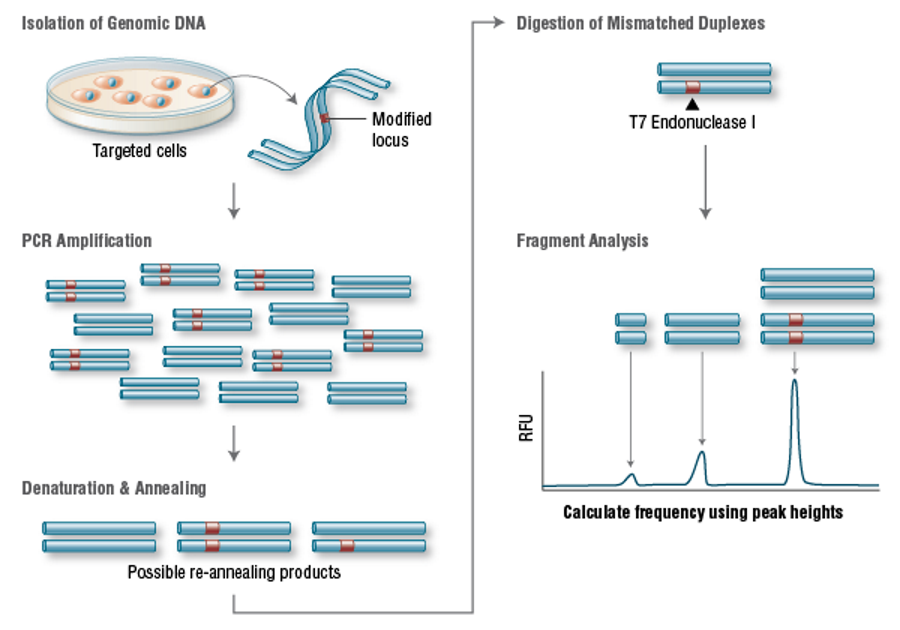
Figure 3. T7 Endonuclease I Targeting Efficiency Assay
[Figure 3:genomic DNA is amplified with primers surrounding the modified site. Then the PCR products were denatured and re-annealed to produce three possible structures. The double strands containing mismatch were digested by T7 endonuclease I. Then the DNA was separated by electrophoresis and fragment analysis was used to calculate the targeting efficiency.]
Another important parameter is the incidence of miss mutations. This mutation is likely to occur in sites where there are only a few nucleotides different from the original sequence, as long as they are adjacent to the PAM sequence. This is because Cas9 can tolerate up to 5 base mismatches in the original spacer region36or single base differences in the PAM sequence38. Off-target mutations are often more difficult to detect and require genome-wide sequencing to completely rule them out.
The CRISPR system has recently been improved to reduce miss mutations by using truncated gRNA (truncated within the crRNA derivative sequence) or by adding two additional guanine (G) nucleotides at the 5 'end28,37. Another way for researchers to try to minimize miss effects is to use "paired incisions"20. The strategy uses D10ACas9 and two sgRNA complementary to adjacent regions on the relative chain of the target site (figure 2-B). Although this induces DSB in the target DNA, it is expected to produce only a single notch at the miss site, resulting in minimal miss mutation. By using computing to reduce miss mutations, several teams have developed web-based tools to facilitate the identification of potential CRISPR targets and assess their potential for miss cutting. Examples include CRISPR design tools38and ZiFiTTargeter, version 4.239,40.
Application as a tool for genome editing and genome targeting
The CRISPR/Cas9 system has been widely adopted since its first demonstration in 20129. At present, it has been successfully used in many important genes in cell lines and organisms, including human34, bacteria41, zebrafish32, Cryptodermis elegans42, plants34, tropical fish43, yeast44, fruit flies45, monkeys46, rabbits47, pigs42, rats48and mice49. Several teams have now used this approach to introduce single point mutations (deletions or insertions) into a particular target gene through a single gRNA. Replacing with a pair of gRNA-guided Cas9 nucleases can also induce large deletions or genome rearrangements, such as inversion or translocation50. An exciting recent research development is the use of the dCas9 version of the CRISPR/Cas9 system for transcriptional regulation of protein domains26,51,52, epigenetic modification25, and microscopic visualization of specific genomic loci27.
The CRISPR/Cas9 system only needs to redesign the crRNA to change the target specificity. This is in sharp contrast to other genome editing tools, including ZFNs and TALENs,which need to redesign the protein DNA interface. In addition, CRISPR/Cas9 can quickly detect the gene function of the whole genome by generating a large gRNA library51,53 for genome screening.
The future of CRISPR/Cas9
Significant progress has been made in developing Cas9 into a set of tools for cellular and molecular biology research, which may be due to the simplicity, efficiency and versatility of the system. Among the designed nuclease systems that can be used in precision genome engineering, the CRISPR/Cas system is by far the easiest to use. It is clear that the potential of Cas9 is beyond the scope of DNA cleavage, and its usefulness in recruiting genomic site-specific proteins may only be limited by our imagination.
References
1. Cong L., et al. (2013) Science, 339, 819–823.
2. Capecchi, M.R. (2005) Nat. Rev. Genet. 6, 507–512.
3. Fire, A., et al. (1998) Nature, 391, 806–811.
4. Elbashir, S.Mm, et al. (2002) Methods, 26, 199–213.
5. Martinez, J., et al. (2003) Nucleic Acids Res. Suppl. 333.
6. Alic, N, et al. (2012) PLoS One, 7, e45367.
7. Miller, J., et al. (2005) Mol. Ther. 11, S35–S35.
8. Mussolino, C., et al. (2011) Nucleic Acids Res. 39, 9283–9293.
9. Ishino, Y., et al. (1987) J. Bacteriol. 169, 5429–5433.
10. Barrangou, R., et al. (2007). Science, 315, 1709–1712.
11. Jinek, M., et al. (2012) Science, 337, 816–821.
12. Deltcheva, E., et al. (2011) Nature, 471, 602–607.
13. Sapranauskas, R., et al. (2011) Nucleic Acids Res. 39, 9275–9282.
14. Nishimasu, H., et al. (2014) Cell, doi:10.1016/j.cell.2014.02.001
15. Swarts, D.C., et al. (2012) PLoS One, 7:e35888.
16. Sternberg, S.H., et al. (2014) Nature, doi:10.1038/nature13011.
17. Overballe-Petersen, S., et al. (2013) Proc. Natl. Acad. Sci. U.S.A. 110,19860–19865.
18. Gong, C., et al. (2005) Nat. Struct. Mol. Biol. 12, 304–312.
19. Davis, L., Maizels, N. (2014) Proc. Natl. Acad. Sci. U S A, 111, E924–932.
20. Ran, F.A., et al. (2013) Cell, 154, 1380–1389.
21. Qi, L.S., et al. (2013) Cell, 152, 1173–1183.
22. Gasiunas, G., et al. (2012) Proc. Natl. Acad. Sci. U S A, 109, E2579–2586.
23. Maede, M.L., et al. (2013) Nat. Methods, 10, 977–979.
24. Gilbert, L.A., et al. (2013) Cell, 154, 442–451.
25. Hu, J., et al. (2014) Nucleic Acids Res. doi:10.1093/nar/gku109.
26. Perez-Pinera, P., et al. (2013) Nat. Methods, 10, 239–242.
27. Chen, B., et al. (2013) Cell, 155, 1479–1491.
28. Seung, W., et al. (2014) Genome Res. 24, 132–141.
29. Miller, J.C., et al. (2011). Nat. Biotechnol. 29, 143–148.
30. Mussolino, C., et al. (2011). Nucleic Acids Res. 39, 9283–9293.
31. Maeder, M.L., et al. (2008) Mol. Cell, 31, 294–301.
32. Hwang, W.Y., et al. (2013) PLoS One, 8:e68708.
33. Feng, Z., et al. (2013) Cell Res. 23, 1229–1232.
34. Mali, P., et al. (2013) Science, 339, 823–826.
35. Zhou, J., et al. (2014) FEBS J. doi:10.1111/febs.12735.
36. Fu, Y., et al. (2013) Nat. Biotechnol. 31, 822–826.
37. Fu, Y., et al. (2014) Nat Biotechnol. doi: 10.1038/nbt.2808.
38. Hsu, P.D., et al. (2013) Nat. Biotechnol. 31, 827–832.
39. Sander, J.D., et al. (2007) Nucleic Acids Res. 35, W599-605.
40. Sander, J.D., et al (2010) Nucleic Acids Res. 38, W462–468.
41. Pyne M.E., et al. (2015) Appl Environ Microbiol 81:5103–5114.
42. Oh J.H., et al. (2014) Nucleic Acids Res 42:e131.
43. Jiang W., et al. (2013) Nat Biotechnol 31:233–239.
44. Hai, T., et al. (2014) Cell Res. doi: 10.1038/cr.2014.11.
45. Guo, X., et al. (2014) Development, 141, 707–714.
46. DiCarlo, J.E., et al. (2013) Nucleic Acids Res. 41, 4336–4343.
47. Gratz, S.J., et al. (2014) Genetics, doi:10.1534/genetics.113.160713.
48. Niu, Y., et al. (2014) Cell, 156, 836–843.
49. Yang, D., et al. (2014) J. Mol. Cell Biol. 6, 97-99.
50. Ma, Y., et al. (2014) Cell Res. 24, 122–125.
51. Mashiko, D., et al. (2014) Dev. Growth Differ. 56, 122–129.
52. Gratz, S.J., et al. (2013) Fly, 249.
53. Mali, P., et al. (2013) Nat. Biotechnol. 31, 833–838.