My Cart
You have no items in your shopping cart.

INTRODUCTION
Biotechnology fields such as tissue engineering and drug delivery continue to grow with the increasing demand for diverse functional biomaterials. For decades, research in polymeric biomaterials has focused on testing the biocompatibility of polymers developed for other applications or their processing (e.g.: electrospinning, solvent casting/porogen leaching, 3D printing). More recently, researchers have turned their attention to the synthesis of materials specifically for biomedical use, including the synthesis of proteins, sugar mimetics, and polymers compatible with aqueous media, as well as the chemical modification of natural polymers (e.g., gelation to increase the stability of substances in vivo). Over the past decade, polymer chemists have created a promising scenario for designing biomaterials for use as cell scaffolds or drug delivery.
Hydrogel is one of the most concerned biological materials for a long time.1 Because of its chemical and physical properties are very close to the natural environment of cells, it has been widely studied as a 2D and 3D scaffold for cells.2 Hydrogels can be formed from synthetic (e.g., poly(ethylene glycol), poly (hydroxyethyl methacrylate)) and naturally occurring polymers (e.g., collagen, hyaluronan, heparin), and are useful 3D models of tissue culture due to their high water content and ability to form in the presence of cells, proteins and DNA. Depending on the reactivity of the constituent materials, gelation can be induced using pH3, temperature4, coulombic interactions, covalent bonding, non-covalent interactions5, or polymerization.
PEG
Polyethylene glycol (PEG) is a hydrophilic polymer that can have a high water content when forming a cross-linked network. PEG is a suitable material for biological applications because it usually does not elicit an immune response.
6 Since the 1970s, PEG has been used to modify therapeutic proteins and peptides to increase their solubility, lower their toxicity and to prolong their circulation half-life.7 Researchers have been experimenting with using PEG hydrogels for cell culture since the late 1970s. Polyethylene glycol hydrogels have good chemical reactivity, and a variety of reactive groups can be used to form hydrogels and achieve efficient chemical modification.PEG MACROMERS
In order to form a hydrogel, PEG must be cross-linked. Initially, PEG was cross-linked non-specifically using ionizing radiation.8 PEG hydrogels are now typically synthesized via covalent cross-linking of PEG macromers with reactive chain ends.
PEG macromers with reactive chain ends such as acrylate, methacrylate, allyl ether, maleimide, vinyl sulfone, NHS ester and vinyl ether groups (Figure 1) are easily synthesized from readily available starting materials. The alcohol chain ends of PEG can be esterified using acid chlorides (e.g., acryloyl chloride, methacryloyl chloride) in the presence of base. PEG chain ends can be etherified under basic conditions by reaction with alkyl halides such as 2-chloroethyl vinyl ether or allyl bromide. PEG divinyl sulfone is prepared by coupling PEG to a large excess of divinyl sulfone or by a multistep process to prepare chloroethyl sulfone chain ends that undergo basic elimination to form vinyl sulfone groups9.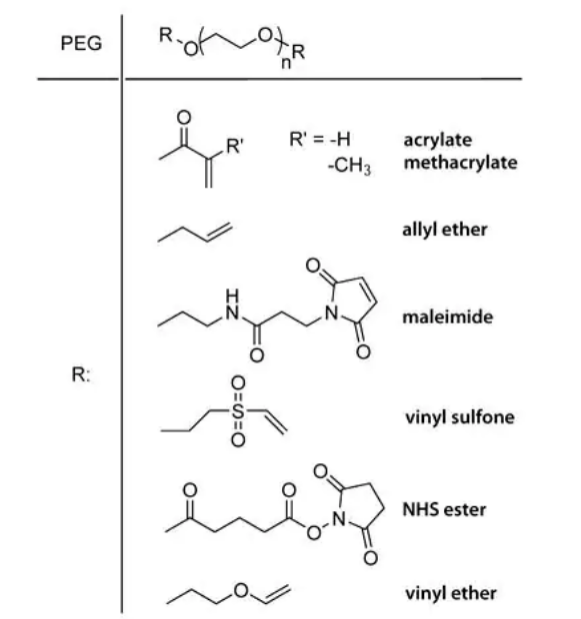
Figure 1. End groups of different PEG macromers.
The two ends of the polymer can be the same or different functional groups. Homodifunctional polymers are commonly used to form networks, while heterodifunctional polymers can be used to link small molecules with therapeutic properties into hydrogel networks.
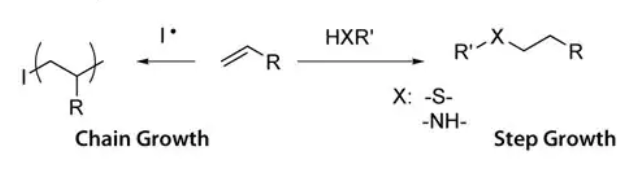
Figure 2. Chain growth and step growth reactions.
The mesh structure that results from different mechanisms is depicted in Figure 3. In chain growth networks, a kinetic chain is formed at the crosslink site, while in step growth networks, the crosslink sites bear the same functionality as the multifunctional cross-linker, neglecting defects. In both chain and step growth, network defects such as loops, permanent entanglements and dangling chain ends may exist. The chemical identity of the macromer and the mechanism of hydrogel formation are both important as each influences the cross-link density of the hydrogel network. Material properties that are important to 2D and 3D culture are easily controlled through the chemistry of hydrogel formation. As cross-linking density increases, mesh size decreases, swelling ratio decreases, and storage modulus increases. Varying the molecular weight of the PEG macromer results in coarse control over hydrogel properties (large differences in cross-linking density). Varying the reaction mechanism used to produce the hydrogels results in fine control over hydrogel properties (can be used to tune cross-linking density of a system).
In order to use 3D hydrogel scaffolds to study cell differentiation and tissue evolution, it is critical to be able to control the physical and chemical properties of the gel in a spatially and temporally controlled manner.10 Polymeric material properties are typically changed through polymerization/cross-linking (bond forming events) or through controlled degradation and/or release (bond breaking events). Bond forming events typically often use small molecule reagents (initiators, catalysts, monomers, ligands to be conjugated to the material) while bond breaking typically does not rely on exogenous reagents. Small molecules often have more adverse effects in vitro and in vivo than polymeric reagents so many research groups use degradation as a tool for in situ manipulation of polymeric biomaterials.
HYDROLYTIC DEGRADATION
Alcohol chain ends on PEG can initiate ring-opening reactions of 3,6-dimethyl-1,4-dioxane-2,5-dione and 1,4-Dioxane-2,5-dione to generate PEG-lactide and PEG-glycolide, respectively (Figure 4).11 The ring-opening reaction is typically catalyzed by tin(II)-2-ethylhexanoate,12 although the reaction is also easily accomplished using dimethylaminopyridine as a catalyst,13 which may be easier to remove than the residual tin. The alcohol chain ends of PEG-lactide or PEG-glycolide are easily functionalized with reactive double bonds such as acrylate and methacrylate.
MMP-degradable linkages have also been used to tether therapeutic agents into hydrogels. For example, growth factors such as vascular endothelial growth factor (VEG-F) can be released via enzymatic degradation of an MMP-sensitive tether to induce angiogenesis.16
In both hydrolysis and enzymolysis, the rate of degradation is predetermined by the chemistry of the macromer. In hydrolysis, the degradation rate of the material is pre-engineered through the identity (e.g., hydrophobicity or hydrophilicity) and number of the hydrolysable groups, and cannot be changed once the material is fabricated. In enzymolysis, the degradation typically occurs in an area local to the cells producing the enzyme. While hydrolysis and enzymolysis are both effective methods for sustained hydrogel degradation and sustained release of therapeutic agents, the rate of release cannot be adjusted or arrested after the hydrogel is fabricated, and release is not spatially controlled.
Figure 5. Enzymatically degradable hydrogels via Michael addition of cysteinecontaining peptides to vinyl sulfone groups.
PHOTODEGRADABLE HYDROGELS
In contrast to hydrolytically and enzymatically degradable linkages, photodegradable linkages allow precise spatial and temporal control over degradation and release. While many researchers have reported photopolymerizable hydrogels, and photofunctionalizable hydrogels, very few reports exist of biocompatible photodegradable hydrogels. Kloxin and Kasko reported photodegradable hydrogel networks formed from 2-methoxy-5-nitro-4-(1-hydroxyethyl) phenoxybutanoate-containing PEG macromers (Figure 6)17; the photodegradation behavior of the ortho-nitrobenzyl (o-NB) linker group is well-characterized. Hydrogels formed from the photodegradable macromer show bulk degradation upon exposure to light that is dependent on exposure time, wavelength, and light intensity. When the light is shuttered, degradation is arrested; the sample continues photolyzing once light exposure resumes. hMSCs (human mesenchymal stem cells) encapsulated in a hydrogel containing the photo-releasable cell-adhesive ligand RGDS (Arg-Gly-Asp-Ser) differentiate down the chondrogenic pathway when the RGD is released at day ten (corresponding to the downregulation of fibronectin during chondrogenesis). Surface erosion and through-gel lithography of this degradable hydrogel can be used to form features over a range of lengths scales, from 10-7 m to 10-2 m or larger.18 Partial degradation in a local area results in decreased cross-link density and increased swelling, providing a means to etch softer features onto a hydrogel that protrude out from the gel.
In addition to single photon photolysis, the o-NB containing hydrogels are also susceptible to two-photon photolysis, allowing for 3D etching.19-20 In single photon reactions, any area exposed to the light will react. In contrast, multi-photon lithography should occur only where multiple photons are simultaneously absorbed, which occurs at the focal volume of the light source (Figure 7). Typical wavelengths in single photon lithography of biomaterials range from long wave UV (≥365 nm) into the visible region, while two-photon lithography uses IR light (typically ~740–800 nm). IR light is more biocompatible and less destructive to live tissues and offers greater penetration depth. The probability of twophoton absorption occurring is also tightly limited to the focal point of the focused light, rather than along the entire path of the light, providing 3D control over excitation. Both single- and multi-photon reactions have the potential to pattern materials with features smaller than 500 nm, much smaller than the size of a mammalian cell.21 This represents an unprecedented level of spatial control over hydrogel scaffold structure and chemistry.
The o-NB linker can also be used to tether therapeutic agents into hydrogels for delivery to live cells. Griffin et al. demonstrated the controlled release of fluorescein tethered into a hydrogel through an o-NB-PEG macromer.22 The release of this model therapeutic as a function of light exposure at multiple wavelengths (365-436 nm), intensities (5-20 mW/cm2) and durations (0-20 minutes) was quantified. While the fastest release occurs at 365 nm (which corresponds to a higher molar absorptivity of the o-NB linker at that wavelength), significant release is also seen at 405 nm; the release is easily modeled from physical constants of the molecules (such as molar absorptivity). Light attenuation allows the facile formation of chemical and mechanical gradients in these systems.
References
1. Drury JL, Mooney DJ. 2003. Hydrogels for tissue engineering: scaffold design variables and applications. Biomaterials. 24(24):4337-4351. http://dx.doi.org/10.1016/s0142-9612(03)00340-5
2. Lee KY, Mooney DJ. 2001. Hydrogels for Tissue Engineering. Chem. Rev.. 101(7):1869-1880. http://dx.doi.org/10.1021/cr000108x
3. HOFFMAN AS. Hydrogels for Biomedical Applications. 944(1):62-73. http://dx.doi.org/10.1111/j.1749-6632.2001.tb03823.x
4. Hoffman AS. 2002. Hydrogels for biomedical applications. Advanced Drug Delivery Reviews. 54(1):3-12. http://dx.doi.org/10.1016/s0169-409x(01)00239-3
5. Tibbitt MW, Anseth KS. 2009. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng.. 103(4):655-663. http://dx.doi.org/10.1002/bit.22361
6. Lin C, Anseth KS. 2009. PEG Hydrogels for the Controlled Release of Biomolecules in Regenerative Medicine. Pharm Res. 26(3):631-643. http://dx.doi.org/10.1007/s11095-008-9801-2
7. Richter A, Paschew G, Klatt S, Lienig J, Arndt K, Adler H. Review on Hydrogel-based pH Sensors and Microsensors. Sensors. 8(1):561-581. http://dx.doi.org/10.3390/s8010561
8. Ruel-Gariépy E, Leroux J. 2004. In situ-forming hydrogels?review of temperature-sensitive systems. European Journal of Pharmaceutics and Biopharmaceutics. 58(2):409-426. http://dx.doi.org/10.1016/j.ejpb.2004.03.019
9. Yang Z, Xu B. 2007. Supramolecular hydrogels based on biofunctional nanofibers of self-assembled small molecules. J. Mater. Chem.. 17(23):2385. http://dx.doi.org/10.1039/b702493b
10. Zalipsky S, Harris JM. 1997. Introduction to Chemistry and Biological Applications of Poly(ethylene glycol).1-13. http://dx.doi.org/10.1021/bk-1997-0680.ch001
11. Davis FF. 2002. The origin of pegnology. Advanced Drug Delivery Reviews. 54(4):457-458. http://dx.doi.org/10.1016/s0169-409x(02)00021-2
12. Morpurgo M, Veronese FM, Kachensky D, Harris JM. 1996. Preparation and Characterization of Poly(ethylene glycol) Vinyl Sulfone. Bioconjugate Chem.. 7(3):363-368. http://dx.doi.org/10.1021/bc9600224
13. Lutolf MP, Hubbell JA. 2005. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat Biotechnol. 23(1):47-55. http://dx.doi.org/10.1038/nbt1055
14. Sawhney AS, Pathak CP, Hubbell JA. 1993. Bioerodible hydrogels based on photopolymerized poly(ethylene glycol)-co-poly(.alpha.-hydroxy acid) diacrylate macromers. Macromolecules. 26(4):581-587. http://dx.doi.org/10.1021/ma00056a005
15. Du YJ, Lemstra PJ, Nijenhuis AJ, Van Aert HAM, Bastiaansen C. 1995. ABA Type Copolymers of Lactide with Poly(ethylene glycol). Kinetic, Mechanistic, and Model Studies. Macromolecules. 28(7):2124-2132. http://dx.doi.org/10.1021/ma00111a004
16. Kim H, Kim HW, Suh H. 2003. Sustained release of ascorbate-2-phosphate and dexamethasone from porous PLGA scaffolds for bone tissue engineering using mesenchymal stem cells. Biomaterials. 24(25):4671-4679. http://dx.doi.org/10.1016/s0142-9612(03)00358-2
17. Benoit DS, Nuttelman CR, Collins SD, Anseth KS. 2006. Synthesis and characterization of a fluvastatin-releasing hydrogel delivery system to modulate hMSC differentiation and function for bone regeneration. Biomaterials. 27(36):6102-6110. http://dx.doi.org/10.1016/j.biomaterials.2006.06.031
18. West JL, Hubbell JA. 1999. Polymeric Biomaterials with Degradation Sites for Proteases Involved in Cell Migration. Macromolecules. 32(1):241-244. http://dx.doi.org/10.1021/ma981296k
19. Lutolf MP, Hubbell JA. 2003. Synthesis and Physicochemical Characterization of End-Linked Poly(ethylene glycol)-co-peptide Hydrogels Formed by Michael-Type Addition. Biomacromolecules. 4(3):713-722. http://dx.doi.org/10.1021/bm025744e
20. Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. 2003. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: Engineering cell-invasion characteristics. Proceedings of the National Academy of Sciences. 100(9):5413-5418. http://dx.doi.org/10.1073/pnas.0737381100
21. Zisch AH, Lutolf MP, Ehrbar M, Raeber GP, Rizzi SC, Davies N, Schmökel H, Bezuidenhout D, Djonov V, Zilla P, et al. 2003. Cell-demanded release of VEGF from synthetic, biointeractive cell-ingrowth matrices for vascularized tissue growth. FASEB j.. 17(15):2260-2262. http://dx.doi.org/10.1096/fj.02-1041fje
22. Kloxin AM, Kasko AM, Salinas CN, Anseth KS. 2009. Photodegradable Hydrogels for Dynamic Tuning of Physical and Chemical Properties. Science. 324(5923):59-
63. http://dx.doi.org/10.1126/science.1169494