My Cart
0
You have no items in your shopping cart.
Nucleic
acid electrophoresis is used in many molecular biology research applications to
test experimental results, and sometimes to isolate and purify samples for
subsequent applications. Therefore, the application of conventional nucleic
acid electrophoresis can generally be divided into analytical and preparative
types, both of which rely on separation, dissolution, and quantification
techniques. These applications include:
1. Analytical electrophoresis
applications
a. PCR, restriction digestion, ligation, colony
screening, reverse transcription, in vitro transcription
b. Gel quantitation
c. Nucleic acid purification, sample fragmentation,
oligo synthesis efficiency
d. Southern blot, northern blot, nuclease protection
assay
e. DNA conformation, EMSA
2. Preparative electrophoresis applications
a. PCR, restriction digestion, oligonucleotide
synthesis, NGS size selection
b. Gel purification
i. Agarose gels
ii. Polyacrylamide gels
1. Analytical electrophoresis used to
confirm experimental results
The
results can be examined using analytical nucleic acid electrophoresis before
proceeding to the next workflow or another set of experiments. As described
below, the method is primarily evaluated for the presence or absence of the
target band in the gel, as well as the strength, migration pattern, mobility,
and hybridization of the band.
Electrophoretic analysis of nucleic acids is usually performed immediately after the following techniques (Figure 1) to determine the success and efficiency of the experiment:
lIn a few hours, polymerase chain reaction, or PCR, amplifies one copy of the target sequence to millions of copies. Electrophoresis was performed after end-point PCR to confirm the amplification of the target and its yield.In a few hours, polymerase chain reaction, or PCR, amplifies one copy of the target sequence to millions of copies. Electrophoresis was performed after end-point PCR to confirm the amplification of the target and its yield.
lAfter a restriction digestion reaction with a restriction enzyme to cleave a specific sequence on a DNA substrate, a sample is run on a gel to determine the pattern of DNA cleavage (and thus the degree of completion of digestion).
lIn molecular cloning, fragments of DNA are inserted into a vector by a process called ligation. In some cases, electrophoresis may be performed after ligation to assess reaction efficiency (FIG. 2). The ligation products are then used to transform competent cells of cloned organisms, such as E. coli, for propagation, and the resulting colonies are screened to determine whether they carry a vector with the desired insert. PCR and restriction digestion, both of which typically involve electrophoresis as part of the workflow, are commonly used techniques in colony screening.
lReverse transcription is the synthesis of DNA complementary to an RNA template (cDNA) by an enzyme called reverse transcriptase. After cDNA synthesis and removal of the RNA template, the product can be run on denaturing gels to assess reaction efficiency. This method is most commonly used for reverse transcription of RNA of known sequence or length.
lIn vitro transcription is the synthesis of RNA from a DNA template by RNA polymerase. After removal of the DNA template, the resultant RNA can be electrophoresed on denaturing gels to confirm successful transcription. 
Figure 1. Common molecular biological applications of electrophoresis to confirm the success of experiments.
Figure 2. Ligation efficiency determined by gel electrophoresis. λDNA is first cleaved with HindIII, a type II site-specific restriction enzyme (lane 1). Samples were then reattached and analyzed by gel electrophoresis (lane 2); Fully connected DNA is the most prominent band.
b. Nucleic acid samples were
quantified by gel electrophoresis
Electrophoresis
can be used to quantify DNA or RNA bands of interest by means of a standard or
Ladder of known content for each segment. In the quantitative method, the
commonly used spectrophotometric quantitative method can be affected by
pollutants such as nucleotides and primers. However, electrophoresis can
separate target samples from these pollutants and thus become a reliable
quantitative method.
Gel
quantification was performed to estimate the number of samples by comparing the
strength of a band in the sample to a band of similar size in the Ladder
(Figure 3A). Ideally, when using a Ladder designed for quantification,
different numbers of Ladders should be loaded to create a standard content
versus strength curve for more accurate quantification (Figure 3B). In
addition, staining nucleic acids with fluorescent dyes with high sensitivity
and wide dynamic range can further improve gel quantification. Some gel imagers
are equipped with analysis software for simpler quantitative analysis of
samples in the gel, while others have cloud connectivity for data storage.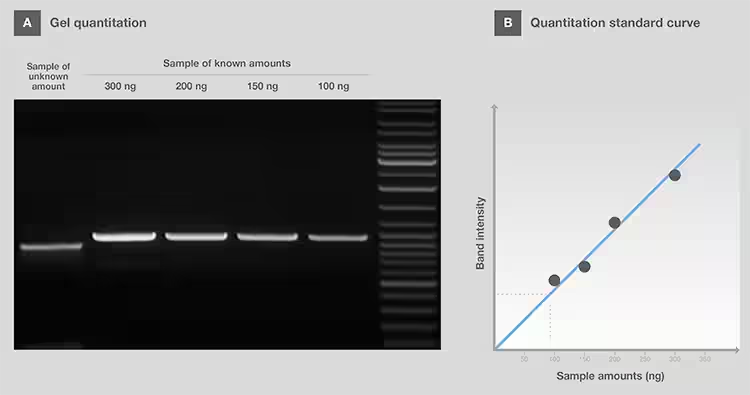
Gel electrophoresis can be used to assess the purity and integrity of nucleic acid samples extracted from their source, as well as the success of sample fragmentation and the percentage of full-length oligonucleotides after synthesis.
l Genomic DNA from contaminated RNA samples, and vice versa, can be tested for sample purity using gel electrophoresis (FIG. 4A). The detection of contamination type depends on the sensitivity of the nucleic acid stain used and the contaminant content.
l Using gel electrophoresis, the integrity of total RNA after extraction can be checked by assessing the relative intensities of 28S and 18S rRNA, where the 2:1 ratio indicates intact RNA. The presence of band tails, especially at lower molecular weights, indicated RNA degradation (FIG. 4B).
l Some protocols require fragmentation of nucleic acid samples, such as when preparing sample inputs for chromatin immunoprecipitation (ChIP) and next-generation sequencing (NGS), to obtain the appropriate fragment size for the next step. The efficiency of sample fragmentation can be detected by gel electrophoresis (FIG. 4C).
l After synthesis, due to With 100% synthetic coupling efficiency, the oligonucleotide product will contain full-length as well as truncated or disabled sequences (i.e., n-1, n-2, etc.)} .
l Electrophoresis can distinguish full-length products from failed sequences according to the size and conformation of oligonucleotides of different lengths.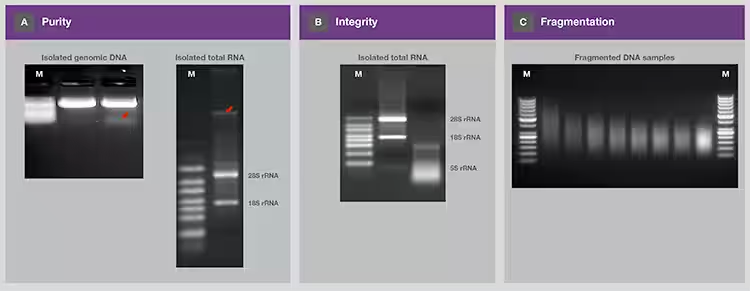
Figure 4.
Determination of sample purity, integrity, and fragmentation by gel
electrophoresis. (A) The extracted genomic DNA and total RNA were analyzed on
separate gels. The red arrows represent contaminating RNA and genomic DNA,
respectively. Contaminating RNA was detected only at the beginning of the
electrophoresis run (≤5 min). (B) The integrity of two samples of purified RNA
was assessed by electrophoresis and analysis of 28S and 18S rRNA. (C) The
efficiency of DNA fragmentation and the distribution of fragmented DNA were
determined on the gel. (M= molecular weight standard. RNA samples were run on denaturing
gels.)
d. Detection of target sequences in
mixed samples
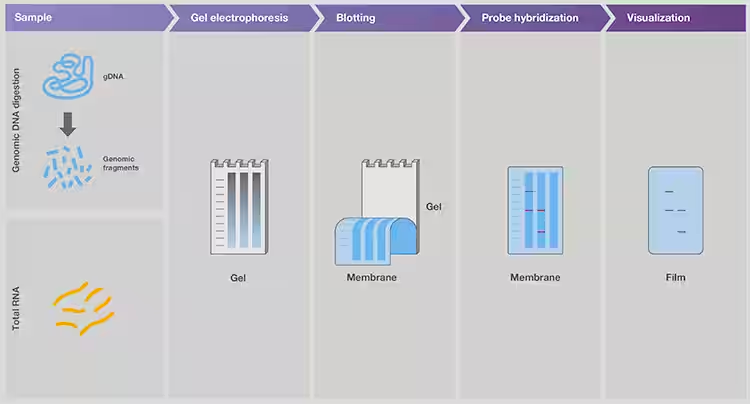
Gel electrophoresis is a key part of the workflow for detecting target sequences in nucleic acid libraries by probe hybridization (Figs. 5, 6). A probe is a single-stranded nucleic acid with a known sequence that is specialized for binding to the target sequence by base complementation.
For
RNA fragment analysis, northern blotting and nuclease protection assays (NPA)
also rely on probe hybridization to detect sequences of interest. The northern
blot followed the same workflow as the Southern blot, except that the input was
RNA (Figure 5). In the ribonucnitase protection assay (RPA), the RNA probe
binds to the target sequence in the sample mixture. The remaining
single-stranded RNA, such as the unbound probe and template and its protrudate
ends, is later digested by ribonucrenases such as RNase A/T1 mix. The bound
probe and sample are subjected to gel electrophoresis for downstream detection
and analysis of the target.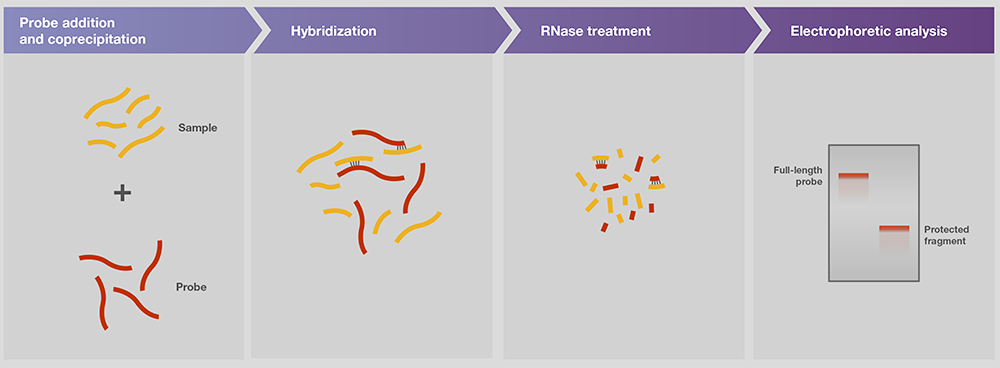
Figure 6. Nuclease protection assay.
e. Assessment of DNA conformation and
nucleic acid-protein complexes
As
discussed in the Electrophoresis Considerations section, different
conformations of plasmid DNA with the same sequence may display different
electrophoretic mobility. This signature can be used to assess DNA
conformation, as well as the level of intact plasmids after extraction.
The electrophoretic migration rate of nucleic acid fragments bound to proteins is slower than that of unbound fragments, formally known as electrophoretic mobility change analysis (EMSA). Methods based on this principle are often referred to as gel shift or gel arrest assays, because bound proteins shift or delay the migration gel of nucleic acid fragments (FIG. 7). Therefore, the electrophoresis step of the experiment can "snap" the balance of bound and free DNA in the sample. For EMSA, low ionic strength buffers should be used in gel preparation and electrophoresis to help stabilize nucleic acid-protein complexes during electrophoresis.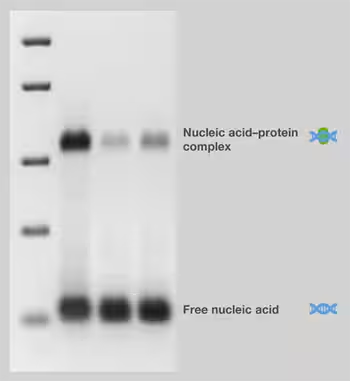
Figure 7. Electrophoretic mobility test.
2. Preparative electrophoresis for the purification of nucleic acid samples
Preparative
application of nucleic acid gel electrophoresis refers to the purification and
extraction of isolated nucleic acid from gel matrix after electrophoresis
analysis. Therefore, such electrophoresis for gel purification is often used as
a preparation step for downstream applications. In addition, the gel
preparation method can be used in conjunction with the analytical method.
Preparative
electrophoresis is commonly used after a variety of molecular biological
techniques and applications, the most common are: PCR; Restriction digestion;
Oligonucleotide synthesis, and the fragmentation, end modification, and
ligation steps of next-generation sequencing (NGS).
PCR products can be purified from gels and DNA that limits enzymatic digestion can be used for downstream applications such as end modification, ligation, cloning and sequencing.
Following
oligonucleotide synthesis, polyacrylamide gel electrophoresis is one of the
main methods for the separation and purification of full-length oligonucleotides
from salt and truncated sequences.
For platforms, samples were fragmented, end-modified and ligated to adaptors as part of sequencing library preparation. After these steps, DNA fragments of a specific size range (for example, 200 to 300bp) are purified in a process known as size selection to remove not only the low and high molecular weight fragments, but also the adaptors, enzymes, and reagents from the previous steps. Fragment screening helps ensure that input samples have fragments of uniform length, thus achieving high-quality and consistent sequencing [1]. Gel electrophoresis is a method of size selection because of the efficiency of selecting segments of specific size in a narrow range (FIG. 8).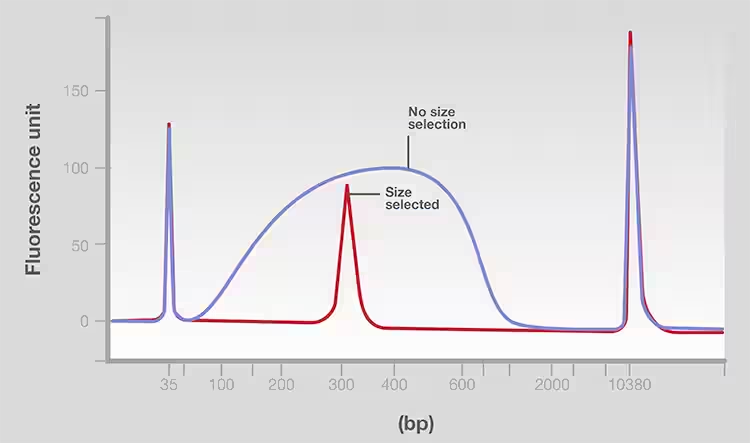
b. Overview of gel Purification
Preparative
electrophoresis is a key step in the separation and purification of nucleic
acids from gels. Nucleic acid purification usually begins by cutting the target
sample band from the gel matrix and then extracting the nucleic acid. When
performing gel purification, the following should be noted:
l To facilitate extraction, the
gel cutting area should be minimized
l Prevent nucleic acid damage
during gel visualization
If
using UV-excitable dyes (e.g., ethidium bromide) for visualization, minimize
the exposure of the sample to radiation, use longer UV wavelengths for
excitation (e.g., 360 nm instead of 254 nm), and choose falling illumination on
transmission. Fluorescent dyes that can be excited by lower energy blue light,
such as SYBR dyes, are better alternatives for sample visualization because
they reduce the radiation damage of the sample.
i. Agarose gel purification
After
cutting the target band from the gel, the agarose can be melted by heating. At
1% gel concentration, the melting point of standard agarose is > 90℃ but
low melting point (LMP) agarose > It melts at 65℃. Due to its low
melting point, the use of LMP agarose can improve the integrity and yield of
extracted nucleic acids. For a gentler method than heating, the enzyme agarose
may be considered to break down the agarose chain into oligosaccharides.
Agarase digestion requires the use of LMP agarose so that the agarose enzyme
remains active at the low gelation temperature of LMP agarose.
After melting or digestion of agarose, nucleic acids can be extracted using any of the following methods [2]. The first simple and effective method of nucleic acid extraction is phenol extraction combined with ethanol precipitation. Due to the toxicity of phenol and the requirement to treat it as organic waste, the current popular method is to separate nucleic acids by binding them to silica based chromatographic columns in the presence of dissociating agents and then elute them from the columns (FIG. 9).
Another
very simple and fast way to separate specific bands is to use special agarose
gels with two rows of Wells[3]. The sample was loaded onto the top row of
Wells, and as electrophoresis progressed, the desired size of the band was
recovered from the bottom row of Wells. This method is especially suitable for
NGS library fragment preparation and downstream cloning experiments.
ii. Polyacrylamide gel purification
Since polyacrylamide gels are formed by polymerization reactions, the gel matrix cannot be removed by heating alone. Nucleic acids are usually extracted from nucleic acid polyacrylamide gels by "extrude and soak" or electroelution methods[4] (FIG. 10).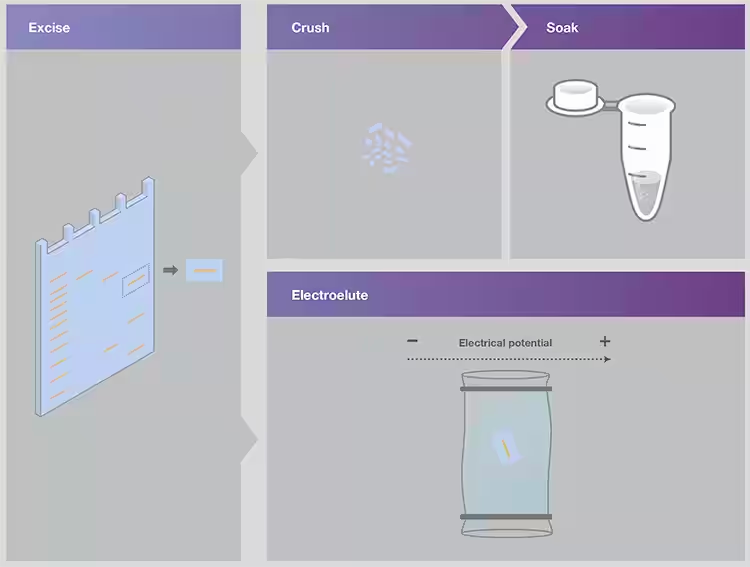
Electroelution methods use an electric field to remove nucleic acids from the gel matrix. A simple method is to place the cut band inside a dialysis membrane with a small molecular weight rejection value. Subsequently, the dialysis membrane containing the gel slice was subjected to electrophoresis to elute the nucleic acid from the gel.
In summary, nucleic acid gel electrophoresis is widely used in various molecular biological workflows and techniques. Although the basic methods of electrophoresis have changed little since the 1970s, it has become recognized as a powerful technique for nucleic acid isolation and analysis in applications ranging from restriction digestion to next-generation sequencing.
Reference:
1. Head SR, Komori HK, LaMere SA et al.(2014) Library
construction for next-generation sequencing: overviews and challenges. Biotechniques, 56(2):61–4, 66, 68.
2. Moore D, Dowhan, D, Chory J et al.(2002) Isolation
and Purification of Large DNA Restriction Fragments from Agarose Gels.In:
Ausubel FM et al.(editors) Current Protocols in Molecular Biology.Supplementary 59: Unit 2.6.
3. Gibson JF, Kelso S, Skevington JH (2010)
Band-cutting no more: A method for the isolation and purification of target PCR
bands from multiplex PCR products using new technology.Mol Phylogenet Evol, 56:1126–1128.
4. Chory J, Pllard JD (1999) Resolution and Recovery of
Small DNA Fragments.In: Ausubel FM et al.(editors) Current Protocols in Molecular
Biology.Supplementary
45: Unit 2.7.
List of related products