My Cart
You have no items in your shopping cart.
Creating an account has many benefits:

Sandra Forbes
Product Manager
Research Background
In the process of developing new drugs, chiral dihydroazaindolecarboxylic acid (R)-1 is in great demand as a key starting material for the synthesis of active pharmaceutical ingredients (APIs). However, the traditional synthesis method, chiral supercritical fluid chromatographic separation, is not only technically difficult but also expensive, which limits the speed and cost control of new drug development to some extent. Therefore, it is particularly urgent to find an economical synthesis process that can replace this expensive method.
Recently, an important breakthrough was achieved by the chemical development work team of Jaehee Lee from Boehringer Ingelheim Pharmaceuticals, Inc. in the United States. Using a second-generation synthetic strategy, they successfully synthesized the complex (R)-2-(4-fluorophenyl)-2-methyl-2,3-dihydro-1H-pyrrolo[2,3-b]pyridine-5-carboxylic acid (R)-1. This new method is not only economically feasible, but also completely eliminates the need to rely on costly chiral supercritical fluid chromatography separations, which can significantly reduce the synthetic cost and provide new, more cost-effective methods for new drug discovery. This new method is not only economically feasible, but also completely independent of expensive chiral supercritical fluid chromatographic separation, thus greatly reducing the synthesis cost and providing a new and more cost-effective synthetic route for new drug discovery. This breakthrough will not only help accelerate the development of new drugs, but may also have a profound impact on the cost control and efficiency improvement of the entire pharmaceutical industry.

In order to address the synthetic challenges encountered in the construction of quaternary stereocenters using asymmetric synthetic methods or chiral starting materials, the researchers have proposed a highly reliable multi-kilogram racemic synthetic scheme. The scheme successfully achieves precise separation of enantiomerically pure substances through an efficient and scalable chemical separation process.
Synthesis Process Development
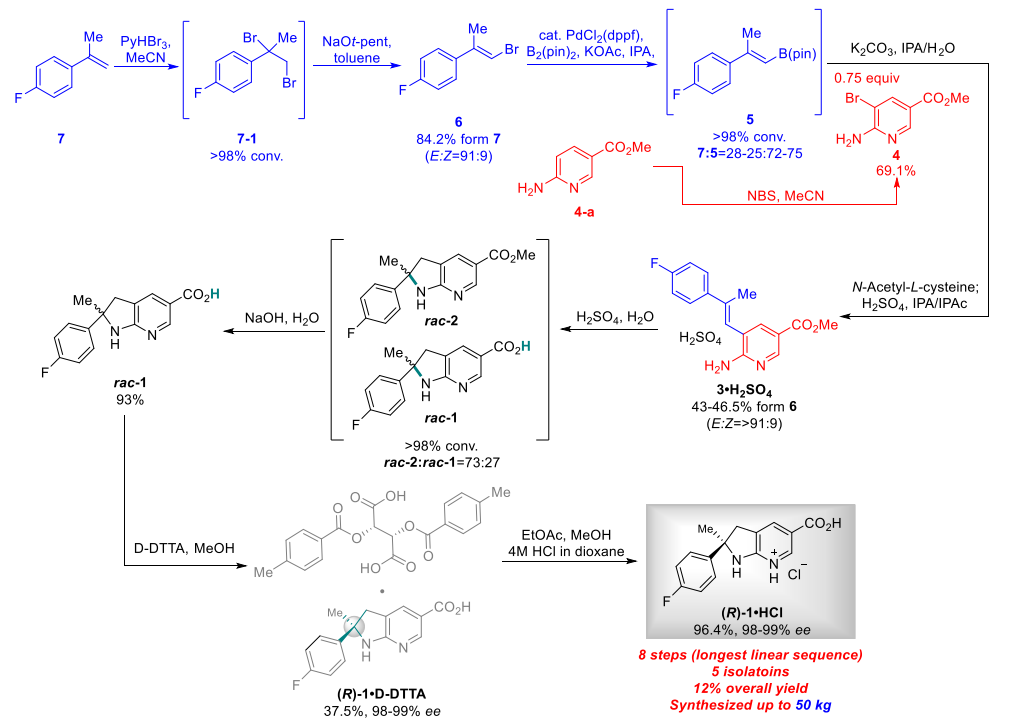
In order to verify the feasibility of synthesizing compound (R)-1 using commercially available starting materials, they first performed preliminary synthesis experiments on a gram scale. In this process, racemic dihydroazaindole rac-2 was prepared via a cross-coupling reaction between 2-amino-3-bromopyridine (A167068) 4 and alkenylboronic acid ester 5. This reaction led to the successful cyclization and assembly of the trisubstituted olefin 3, thus laying a solid foundation for the subsequent synthetic steps.

Initial gram-scale synthesis and kilogram-scale reaction plans
After the formation of the quaternary carbon center (rac-2), the low solubility of rac-2 made the determination of chemical resolution conditions difficult. However, by applying di-p-toluoyltartaric acid (D-DTTA) in the subsequent step (rac-1), they could obtain a high stereoselectivity, which effectively solved this problem.
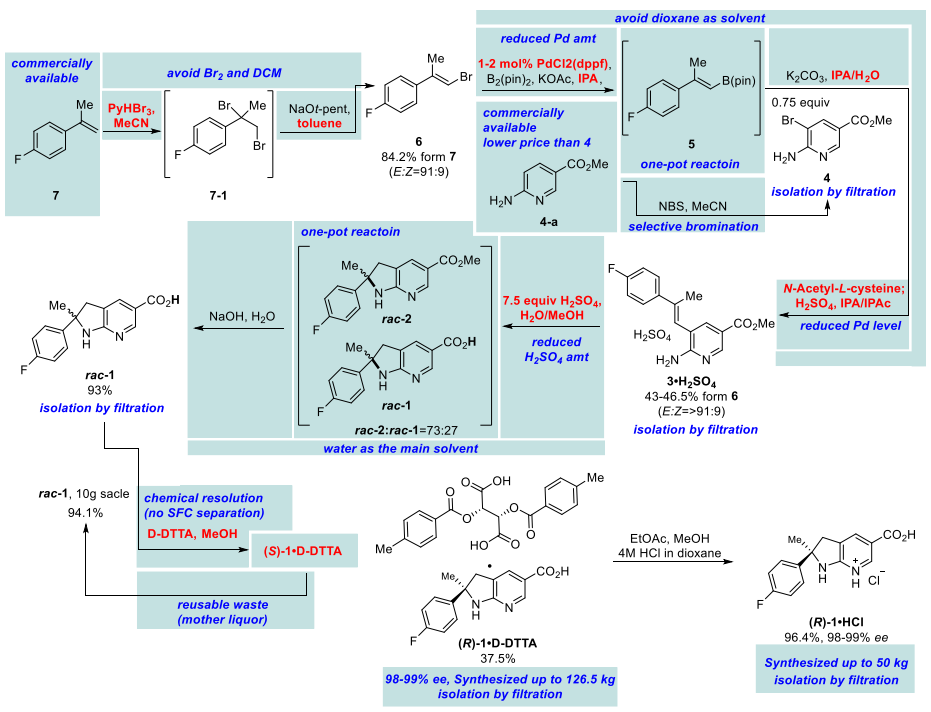
Final process for kilogram scale synthesis
Liquid Br2 poses a handling challenge in large-scale organic synthesis and poses a serious safety hazard due to its high toxicity and corrosiveness. To solve this problem, researchers found that crystalline pyridinium tribromide (pyHBr3) (P160619) can be applied in the preparation of alkenyl bromide as a safer bromination reagent. Compared with liquid bromine (Br2), pyHBr3 is not only easier to handle, but also does not produce by-products. In addition, by using sodium tertiary amyl alcohol solution instead of potassium tert-butoxide solution, they have successfully realized large-scale synthesis, which improves the efficiency as well as ensures the safety of the synthesis process.
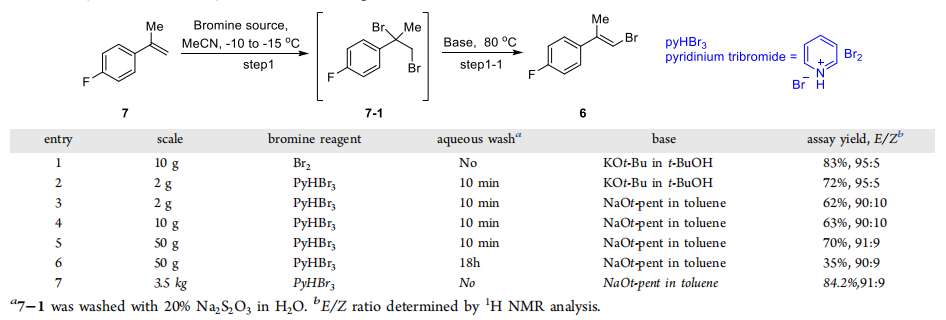
Synthesis of alkenyl bromide 6 by bromination and olefination
In view of the long supply cycle of the starting material, they chose the lower cost and more readily available aminopyridine methyl ester 4-a as an alternative. By brominating aminopyridine 4-a in tetrahydrofuran (THF) and separating it using column chromatography, they achieved product synthesis in high yields. Initially, they tried to isolate the product directly, but suffered large losses. Subsequently, they replaced the solvent with acetonitrile, which significantly reduced the losses. In a large-scale reaction, the use of acetonitrile as a solvent showed consistent results with a small-batch reaction, and the yield was further improved.

Bromination of methyl aminopyridine 4-a
For the initial small-scale process, they employed a 20 mol % Pd catalyst for the borylation and cross-coupling reactions, followed by high purity separation of the products by silica gel chromatography. In order to further optimize the process, reduce the cost and the amount of catalyst used, they decided to use a more efficient single catalyst system that was capable of both borylation and cross-coupling. This improvement not only simplified the process steps but also increased the overall efficiency.
Preparation of trisubstituted olefin 3-acids and their isolation by one-pot method of boronization, Suzuki cross-coupling and salt formation

Preparation of trisubstituted olefin 3-acids and their isolation by one-pot method of boronization, Suzuki cross-coupling and salt formation
In order to mitigate potential risks when using highly acidic reagents for kilo-chemical processing, they conducted small-scale tests with different acids under dilution conditions. Notably, the mother liquor produced during the first resolution process can be efficiently recycled. By racemizing the S-isomer into a mixture of (R)-1 and (S)-1, this mother liquor can be used as a feedstock for another chemical resolution process, thus recycling resources.
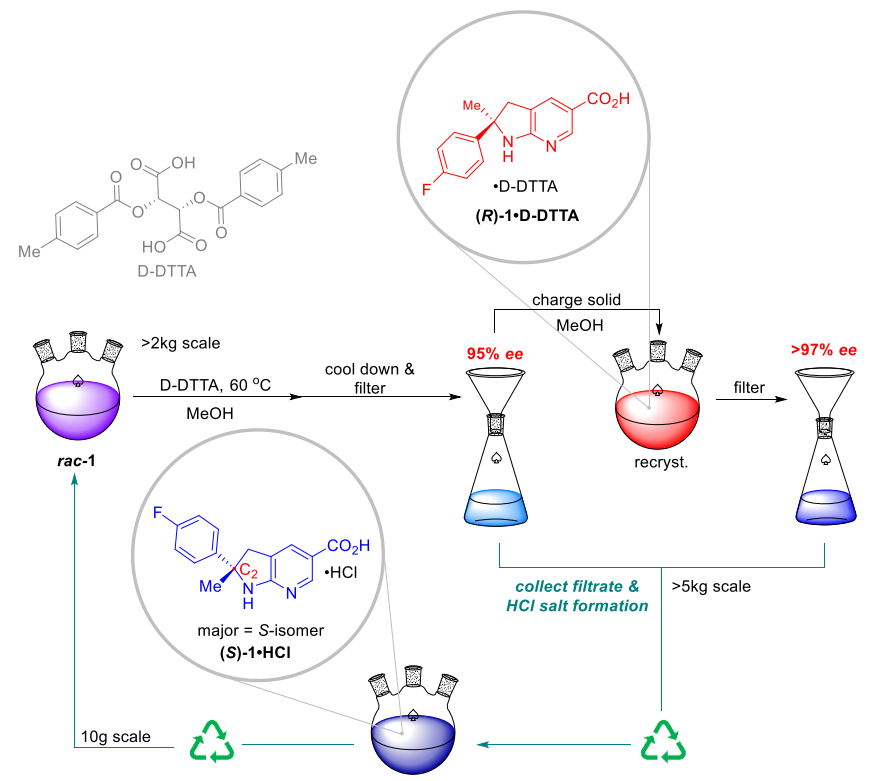
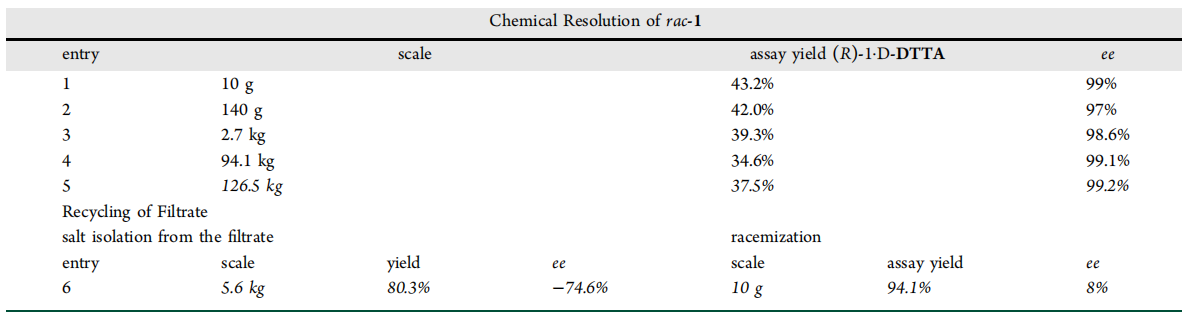
Large-scale chemical splitting and recycling of mother liquor
Although they stirred (R)-1-DTTA in a highly acidic solution, the stereochemical properties were not significantly affected and remained well stabilized because no excess acid was used in the EtOAc/MeOH mixture used as a buffer solvent. This was verified by single-crystal X-ray diffraction, which confirmed the absolute configuration. In addition, the stoichiometry of the methanol molecules in the lattice was found to be 0.25 by measurement.
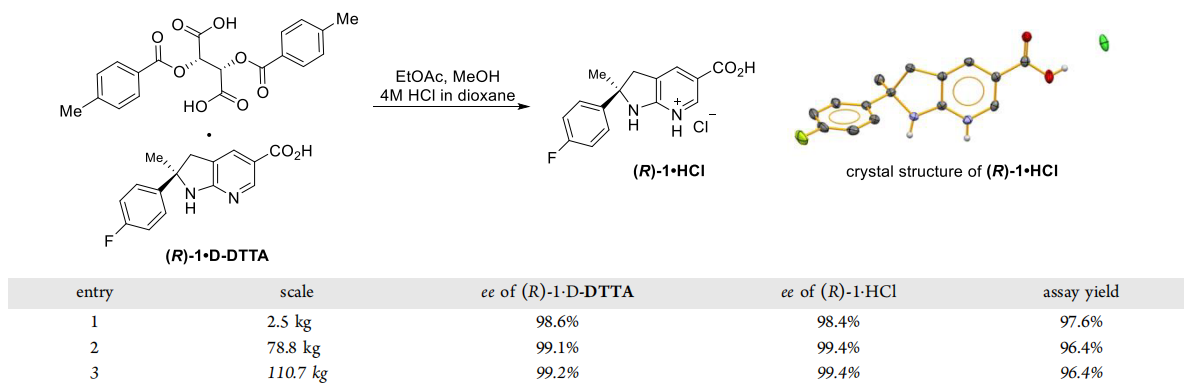
Isolation of (R)-1-HCl from (R)-1-D-DTTA
After the development of a one-pot process, they succeeded in reducing the number of isolation steps, resulting in the efficient synthesis of advanced intermediates. After the critical steps of di-p-toluoyl tartrate breaking and hydrochloric acid formation, they succeeded in isolating the compound (R)-1-HCl, a regulated starting material with high purity.This improvement not only simplified the synthesis process, but also increased the purity of the product, providing a more reliable feedstock for subsequent chemical reactions.
Synthesis of compound (R)-1-HCl
Following a well-designed linear sequence, they successfully synthesized up to 50 kg of compound (R)-1-HCl in 8 steps.This synthesis process excelled in the longest linear sequence with an overall yield of about 12%, achieving high efficiency and feasibility for large-scale production.
Conclusion
This study describes a scalable process for the synthesis of complex regulatory starting materials (R)-1-HCl. Firstly, the construction of quaternary carbon centers was achieved by intramolecular cyclization of compound 3, followed by efficient chemical decomposition using D-DTTA salts, demonstrating excellent stereoselectivity. For future industrial applications, the research team is working on optimizing the process with the aim of reducing the dependence on palladium catalysts and seeking environmentally friendly methods of waste liquid treatment. This study provides a new pathway for the cost-effective and scalable synthesis of dihydroazaindole carboxylic acids, which will be of great value for subsequent drug discovery.
References
1. Jaehee Lee*, Guisheng Li, Xingzhong Zeng et al., Building a Quaternary Stereogenic Center on Dihydroazaindole Carboxylic Acid through Scalable Process Development, Org. Process Res. Dev. 2023, 27, 11, 2045-2056. https://doi.org/10.1021/acs.oprd.3c00231