My Cart
You have no items in your shopping cart.
Basic principles of gel electrophoresis to separate nucleic acids
Gel electrophoresis is a common laboratory technique in molecular biology to identify, quantify, and purify nucleic acids. Because of its speed, simplicity, and versatility, the method is widely employed for separation and analysis of nucleic acids. Using gel electrophoresis, nucleic acids in the range of approximately 0.1–25 kbp can be separated for analysis in a matter of minutes to hours, and separated nucleic acids can be recovered from the gels with relatively high purity and efficiency [1,2].
The technique involves the application of an electrical field to mixtures of charged molecules to cause them to migrate, on the basis of size, charge, and structure, through a gel matrix. The phosphate groups of the ribose-phosphate backbones of nucleic acids are negatively charged at neutral to basic pH (Figure 1A). As such, each nucleotide carries a net negative charge, meaning the overall charge of a nucleic acid molecule is proportional to the total number of nucleotides or its mass. In other words, DNA or RNA molecules carry a constant charge-to-mass ratio. As a result, their mobility in gel electrophoresis is determined mainly on the basis of size when they have comparable structure (learn more about how nucleic acid structure impacts migration). Therefore, when subjected to an electrical field, nucleic acids migrate from the negative electrode (cathode) toward the positive electrode (anode), with shorter fragments moving more rapidly than longer ones, resulting in separation based on size (Figure 1B).

Figure 1. (A) Net negative charges carried by a nucleic acid chain. (B) Separation of nucleic acid fragments of varying lengths in gel electrophoresis.
Furthermore, the migration distances of nucleic acids in gel electrophoresis generally display a predictable correlation with their sizes, enabling calculation of the size of nucleic acids in a given sample. For linear double-stranded DNA fragments, migration distance is inversely proportional to the log of the molecular weight, within a certain range (Figure 2A) [3]. For approximate sizing, migration distances are commonly compared to samples containing molecules of known sizes (molecular weight standards, sometimes referred to as “ladders”) which are often included in the gel run. A widely accepted model of nucleic acid mobility through a gel is “biased reptation”— migration biased towards the applied electrical force and involving a snaking movement where the leading edge pulls the rest (Figure 2B) [4,5]. This model has been visualized by fluorescence microscopy [6].

Figure 2. Mobility of nucleic acids in gel electrophoresis. (A) Correlation of size and migration of linear double-stranded DNA fragments. (B) Biased reptation model.
A brief history of nucleic acid gel electrophoresis
The use of electrophoresis to separate nucleic acids began in the early 1960s. At the time, nucleic acids were commonly fractionated by density gradient centrifugation based on sedimentation velocities, which are determined by size and conformation of the nucleic acids. Density gradient centrifugation took considerable amounts of time, required heavy equipment, and needed high inputs of samples. For alternatives, researchers began to explore characteristics of DNA mobility in ionic, or electrolytic, solutions when an electrical field was applied—a process termed electrophoresis [7,8].
Within a few years, nucleic acid electrophoresis evolved into utilizing a gel matrix as a separation medium, borrowing the technique already employed in protein electrophoresis. Agar (a naturally derived carbohydrate), agarose (a component of agar), polyacrylamide (a synthetic gel), and agarose-acrylamide composite gels were found to be successful as matrices for DNA and RNA electrophoresis, in the mid- to late 1960s[9-11]. Fractionation results from these early gel electrophoresis experiments showed correlations with sedimentation coefficients, or S values, obtained from density gradient centrifugation, an established nucleic acid separation method at the time. With better understanding and advances in the manufacturing of agarose in the late 1960s, agarose gradually replaced agar as the preferred gel electrophoresis medium[12].

Figure 3. Timeline of early development of nucleic acid gel electrophoresis.
In the 1970s, the use of gel electrophoresis for separation and analysis of nucleic acids became more prevalent with the discovery of restriction enzymes and their application in recombinant DNA technology. Sucrose density gradient centrifugation, a common separation method at the time, involved cumbersome processes and could not adequately distinguish similarly sized DNA fragments from restriction digestion. The viscosity of a solution was used empirically as an indicator of the success of restriction digests, since the digestion of DNA from larger to smaller fragments results in less viscous solutions[13]. Cloning of DNA fragments was revolutionized in 1971 when Danna and Nathans first reported sizing of restriction-digested fragments of SV40 DNA by polyacrylamide gel electrophoresis[14]. Although agarose and agarose-polyacrylamide gels were used for separation of RNA and single-stranded DNA in the late 1960s[15,16], the work to analyze restriction-digested fragments by agarose gel electrophoresis was not published until 1973 (Figure 3)[17,18].
The 1970s also brought a breakthrough in the way nucleic acids were detected in gel electrophoresis. Earlier electrophoresis methods relied on radioactive labeling of nucleic acids for visualization of separated molecules. Although highly sensitive, radiolabeling protocols are lengthy and necessitate training in radiation safety. In 1972, two laboratories independently described gel staining with the fluorescent molecule ethidium bromide (EtBr), which became a common and simpler method for detecting nucleic acids with sensitivity of a few nanograms of double-stranded DNA[19-21]. Today, fluorescent stains that are safer and more sensitive and specific than EtBr are available, improving the detection of nucleic acids after gel electrophoresis.
As with the introduction of EtBr, gel electrophoresis became more useful with the advent of “slab” gel formats around 1970, which led to the omnipresent equipment we see today. Earlier studies with gel electrophoresis were performed using tube gels cast into glass tubes of 1–3 mm diameter. It was a very low-throughput method since each tube could accommodate only one sample (Figure 4A). Vertical slab gels (Figure 4B), which were much simpler to prepare and allowed for more samples to be run simultaneously, were first introduced for polyacrylamide in the late 1960s and refined by Studier in the early 1970s[22-23]. The horizontal slab gels for agarose (Figure 4C), similar to the format still used today, was first described by McDonell et al. in 1977[24]. Today, mini precast, ready-to-use gels are available in both polyacrylamide and agarose for safer, faster, and simpler gel electrophoresis. Furthermore, some gel electrophoresis systems take advantage of precast bufferless gels that can be run in as little as 10 minutes. They can also be combined with digital imaging and analysis of the separated nucleic acids for a more efficient and convenient workflow.
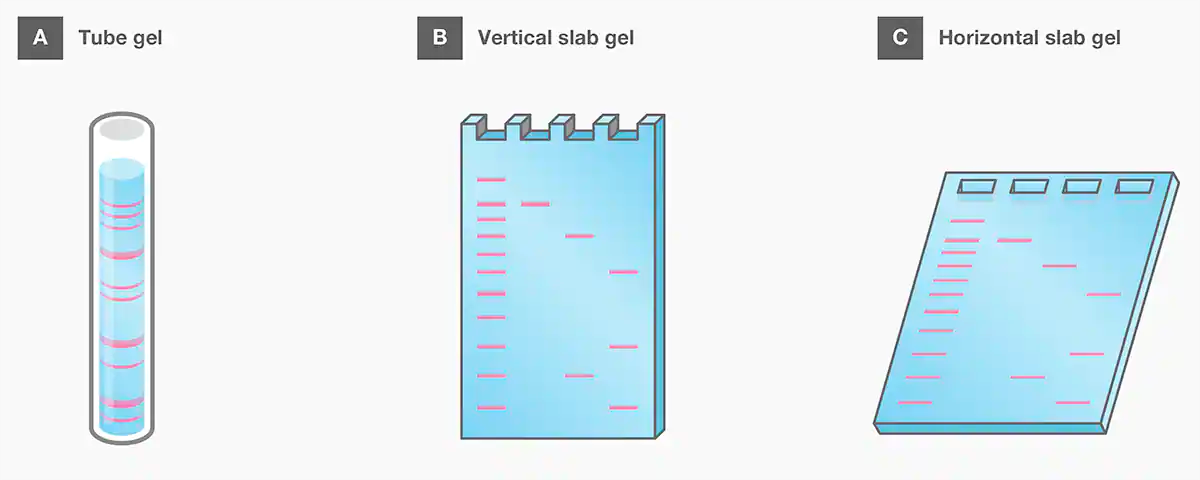
Figure 4. Common gel formats for electrophoresis.
Overall, gel electrophoresis has become a universal technique in molecular biology for separating nucleic acids. Not only is this analytical and preparative method integral to common workflows like molecular cloning and PCR, but it also plays an important role for nucleic acid separation and analysis in emerging technologies like genome editing and next-generation sequencing.
References
18. Aaij C, Borst P (1972) The gel electrophoresis of DNA. Biochim Biophys Acta 269(2):192–200.
20.Borst P (2005) Ethidium DNA agarose gel electrophoresis: how it started. IUBMB Life 57(11):745–747.