Reactions of strained alkenes in click chemistry

Strained alkenes leverage strain relief as a motivating factor, enabling their engagement in click reactions. Trans-cycloalkenes, typically cyclooctenes, and other strained alkenes like oxanorbornadiene, participate in click reactions with various counterparts, including azides, tetrazines, and tetrazoles. These counterparts exhibit specific interactions with the strained alkene, remaining bioorthogonal to the naturally occurring endogenous alkenes present in lipids, fatty acids, cofactors, and other natural products.[1]
Alkene and azide [3+2] cycloaddition
When oxanorbornadiene (or another activated alkene) reacts with azides, it produces triazoles. However, these resulting triazoles lack aromaticity, in contrast to the CuAAC or SPAAC reactions, making them less stable. The activated double bond in oxanobornadiene forms a triazoline intermediate, which subsequently undergoes a spontaneous retro Diels-alder reaction, liberating furan and yielding 1,2,3- or 1,4,5-triazoles. Despite the sluggish nature of this reaction, its utility lies in the relative simplicity of oxabornodiene synthesis. Nevertheless, the reaction is not entirely chemoselective.[2]
Alkene and tetrazine inverse-demand Diels-Alder
Strained cyclooctenes and other activated alkenes undergo an inverse electron-demand Diels-Alder reaction with tetrazines, followed by a retro [4+2] cycloaddition (refer to the figure). [3] Ring strain release, characteristic of trans-cyclooctene reactions, propels this process, making three-membered and four-membered cycloalkenes particularly well-suited alkene substrates due to their elevated ring strain.[3]
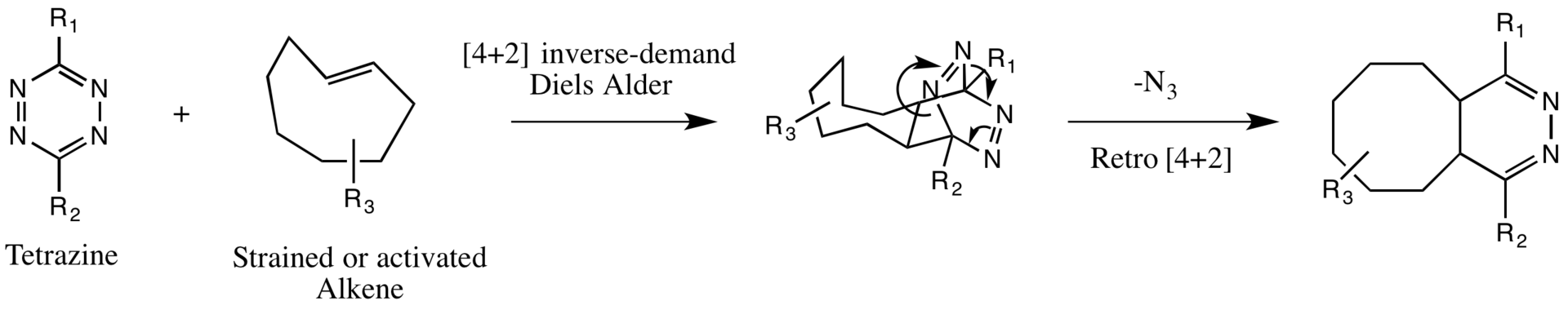
A Tetrazine-Alkene reaction between a generalized tetrazine and a strained, trans-cyclooctene
In line with other [4+2] cycloadditions, the inverse-demand Diels-Alder is facilitated by electron-donating substituents on the dienophile and electron-withdrawing substituents on the diene. The tetrazine, serving as the diene, benefits from additional nitrogens, rendering it a suitable component for this reaction. Additionally, the dienophile, or the activated alkene, is often linked to electron-donating alkyl groups on target molecules, enhancing the suitability of the dienophile for the reaction.[4]
Alkene and tetrazole photoclick reaction
The "photoclick" reaction involving tetrazole-alkene is a dipolar addition method introduced by Huisgen in the late 1960s. Tetrazoles containing amino or styryl groups, activatable by UV light at 365 nm (which does not harm cells), exhibit rapid reactivity (typically within 1–4 minutes) to yield fluorogenic pyrazoline products. This reaction scheme is particularly suitable for live cell labeling, as UV light at 365 nm causes minimal cell damage. Additionally, the reaction proceeds swiftly, allowing brief exposures to UV light. Quantum yields for short-wavelength UV light can exceed 0.5, enabling selective wavelength use when combined with another photoligation reaction. At short wavelengths, tetrazole ligation predominates, while at longer wavelengths, an exclusive reaction (ligation via o-quinodimethanes) takes place. [5]Ultimately, non-fluorogenic reactants result in a fluorogenic product, providing the reaction with an inherent spectrometry handle.
While both tetrazoles and alkene groups have been integrated as protein handles in the form of unnatural amino acids, this advantage is not exclusive. Instead, the photoinducibility of the reaction positions it as a promising candidate for achieving spatiotemporal specificity in living systems. Challenges include the presence of endogenous alkenes, which, although typically cis (as in fatty acids), can still react with the activated tetrazole.[6]
Reference
1. Lang, K.; Chin, J. (2014). "Bioorthogonal Reactions for Labeling Proteins". ACS Chem. Biol. 9 (1): 16-20. https://doi.org/10.1139/cjc-2013-0577
2. (a) van Berkel, S. S.; Dirks, A. T. J.; Meeuwissen, S. A.; Pingen, D. L. L.; Boerman, O. C.; Laverman, P.; van Delft, F. L.; Cornelissen, J. J. L. M.; Rutjes, F. P. J. T. ChemBioChem 2008, 9, 1805. (b) van Berkel, S. S.; Dirks, A. T. J.; Debets, M. F.; van Delft, F. L.; Cornelissen, J. J. L. M.; Nolte, R. J. M.; Rutjes, F. P. J. T. ChemBioChem 2007, 8, 150
3. Liu, Fang; Paton, Robert S.; Kim, Seonah; Liang, Yong; Houk, K. N. (2013). "Diels–Alder Reactivities of Strained and Unstrained Cycloalkenes with Normal and Inverse-Electron-Demand Dienes: Activation Barriers and Distortion/Interaction Analysis". J. Am. Chem. Soc. 135 (41): 15642–15649. https://doi.org/10.1021/ja408437u
4. Rieder, Ulrike; Luedtke, Nathan W. (25 August 2014). "Alkene-tetrazine ligation for imaging cellular DNA". Angew Chem Int Ed Engl. 53 (35): 9168–9172. https://doi.org/10.1002/anie.201403580
5. Menzel, Jan P.; Feist, Florian; Tuten, Bryan; Weil, Tanja; Blinco, James P.; Barner‐Kowollik, Christopher (2019). "Light-Controlled Orthogonal Covalent Bond Formation at Two Different Wavelengths". Angewandte Chemie International Edition. 58 (22): 7470–7474. https://doi.org/10.1016/j.cbpa.2014.05.024
Ramil, Carlo P; Lin, Qing (August 2014). "Photoclick chemistry: a fluorogenic light-triggered in vivo ligation reaction". Current Opinion in Chemical Biology. 21: 89–95. https://doi.org/10.1038/s41570-018-0030-x