My Cart
0
You have no items in your shopping cart.
Creating an account has many benefits:
All-trans retinoic acid (RA, ATRA) is a pleiotropic activation factor that regulates genes associated with normal vertebrate cellular processes such as cell differentiation, cell proliferation, apoptosis, and embryonic development. Retinoic acid and the receptor proteins for which it is an activating ligand have been the focus of numerous papers analyzing retinoic acid-mediated gene expression. Current techniques, including siRNA to knockdown specific genes, RT-PCR to evaluate gene expression, and chromatin immunoprecipitation, have been critical to expand the understanding of the classical RA signaling pathway.
Vitamin A (all-trans-retinol) and its esters are the precursors to functional RA, while carotenes, especially β-carotene, are vitamin A precursors synthesized by plants (see Figures 1, 2, 3). The term “retinoids” encompasses both compounds structurally related to vitamin A, as well as compounds that demonstrate biological vitamin A activity. Animals metabolize dietary carotenes to fat-soluble retinoids and store the retinoids primarily in the liver; animals are unable to synthesize retinoids by other mechanisms. Carotenes are found in several food plants, including yellow, red, or dark green vegetables, and red or yellow non-citrus fruits. Key animal-based dietary sources of vitamin A for humans include liver, fish, and fish oils, such as those from tuna, sardines, and cod. In the United States, dairy products are commonly supplemented with vitamins A and D. After ingestion, vitamin A is bound to retinol binding protein (RBP) for plasma transport and cellular uptake. β-Carotenes and vitamin A esters are chap eroned by chylomicron remnants and transported for uptake by cell surface receptors.1
Intracellular vitamin A levels are maintained by cellular retinol binding proteins (CRBPs) and cellular retinoic acid binding proteins (CRABP-I and CRABP-II).2 CRBPs are associated with retinyl ester storage and transport, while the CRABPs act as retinoic acid chaperones. The endogenous concentration of active retinoid is critical and under close homeostatic control, as either excessive levels of retinoid or retinoid deficiency are teratogenic.2
β-Carotene is cleaved through enzymatic oxidation by β-carotene 15,15’-monooxygenase (EC 1.14.99.36), resulting in two molecules of retinal.3,4The gene sequence of β-carotene 15,15’-monooxygenase has been identified with reasonable homology in a broad variety of species, including human, mouse, chicken, zebrafish, Drosophila, and the cyanobacteria Anabaena variabilis and Synechococcus sp.
Vitamin A and its esters are converted to retinoic acid and related compounds via a two-step oxidation process. Retinols are oxidized to the aldehyde retinal by the action of retinol dehydrogenases (EC 1.1.1.105) or other alcohol dehydrogenases. Retinal is subsequently oxidized to RA by the action of retinal dehydrogenases (EC. 1.2.1.36).2 The cellular retinol-binding protein type I (CRBP-I) acts as a chaperone for retinol and retinal, facilitating their metabolism by oxidating enzymes.
RETINOID NUCLEAR RECEPTOR PROTEINS
Retinoic acid receptors (RAR) and retinoid X receptors (RXR) are the two families of nuclear receptor proteins actively involved in retinoic acid mediated transcription.5 The receptor proteins are members of the nuclear transcription family of steroid/thyroid/ retinoid nuclear receptors. In combination, these nuclear receptors are capable of transcribing and regulating hundreds of genes. RAR and RXR each have three identified isotypes, designated as α, β, and γ. While the gene sequence for each of the RAR isotypes differs significantly from the other two, the sequences for each isotype are highly conserved between humans and mice, leading to the speculation that each RAR isotype has a specific function.6 In addition to the three isotypes, each RAR and RXR have been found to exist as multiple isoforms which result from alternative splicing of a primary transcript or differential promoter usage.7
RAR and RXR receptor proteins are not active in their monomeric forms. RAR does not dimerize with itself but forms heterodimeric proteins with RXR (RAR:RXR). This RAR:RXR protein complex is the predominant receptor complex for retinoic acid signaling. In addition to forming heterodimers with RAR, RXR is able to homodimerize and also dimerize with other receptor proteins, including PPAR.5
The RAR:RXR protein complex binds to retinoic acid response elements (RAREs) or retinoid X response elements (RXREs), each of which are well-defined DNA sequences.7RXR enhances the binding of the RAR protein to the RARE sequences.5 In the absence of ligands at the ligand receptor binding sites, the apo-RAR:RXR heterodimer recruits corepressors, proteins and enzymes that interfere with the transcription process.8The nuclear receptor corepressor (NCoR) and the silencing mediator for retinoid and thyroid hormone receptor (SMRT) inhibit transcription by binding to the ligand-binding domains of RAR and RXR. Protein complexes that contain histone deacetylase activity (HDACs) are subsequently recruited, and these enzymes deacetylate histones within the nucleosome. The loss of acetyl groups increases the positive charge on the histone tails, which increases the attraction of the histone to the negatively charged chromatin. The result is the chromatin becomes more condensed, restricting the ability of transcription factors to attach to the gene initiation site, limiting or preventing transcription (see Figure 4).9
RA and 9-cis-retinoic acid (9-cis-RA) have strong affinities for the ligand receptor site of RAR. 9-cis-RA has a high affinity to the ligand receptor site of RXR, while RA is not a ligand for RXR. While 9-cis-RA has been shown to have specificity for RXR in vitro, there is no strong evidence that 9-cis-RA is the actual ligand involved in the activation of RXR in vivo.1
If a ligand is attached to the ligand-binding domain of the RXR, the conformational change is not sufficient to overcome corepressor recruitment. This inability of RXR activation to initiate the receptor pathway has been called RXR subordination or apo-RAR silencing.5,10 Alternatively, when RA or another retinoid is attached to the ligand-binding domain of the RAR moiety, the protein conformation changes enough to convert the complex from corepressor recruitment to coactivator recruitment. If the binding domains of both RAR and RXR have ligands, the folding of the protein complex is accentuated. RA is transferred from the cytosol to the nucleus by CRABP-II, which acts as a chaperone protein to transport RA and transfer it to the dominant RAR ligand receptor site within the RAR:RXR heterodimer.11
THE CLASSICAL PATHWAY OF RETINOIC ACID-MEDIATED GENE TRANSCRIPTION
The classical pathway of gene expression regulation by RA has four specific steps, although the sequence of the steps is not identical for all genes.7,8 RA-mediated transcription has been defined as having12:
Binding of RA or other retinoid ligand to the ligand binding site
Receptor dimerization (RAR:RXR dimerization)
Binding to DNA (RAREs) by the receptor heterodimer
Transcriptional modulation of the gene, via chromatin remodeling and recruitment of the transcription machinery
DNA binding is directly affected by the action of coactivators and corepressors that modify the chromatin structure of the nucleosome and allow the transcription machinery to access the promoter region of the gene.
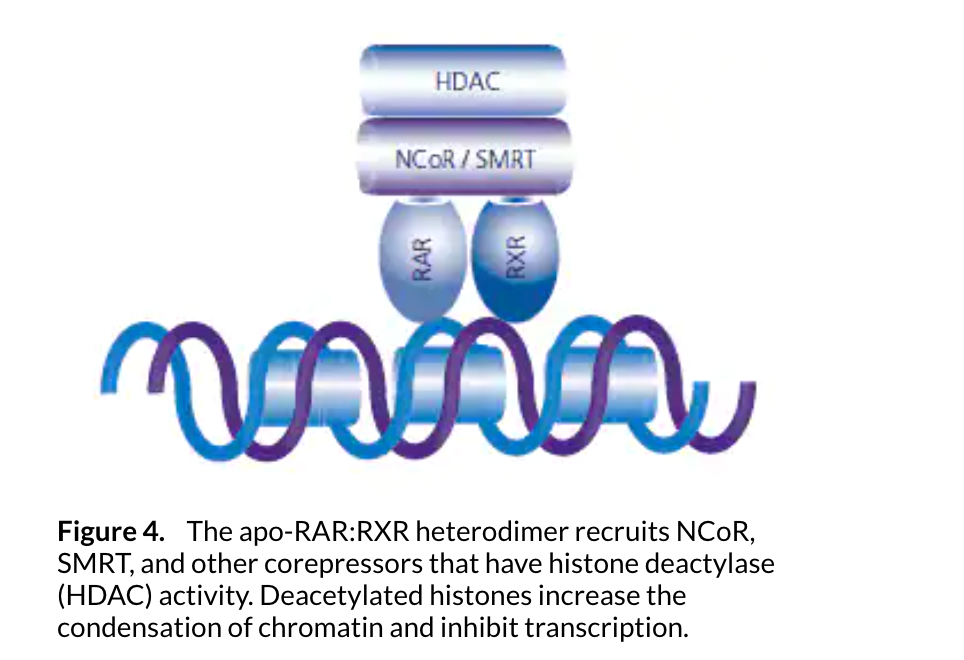
Once retinoid binding modifies the RAR:RXR complex, corepressors are disassociated from the complex and coactivating proteins are recruited. Chromatin remodeling must take place for the transcription machinery complex to access the genes to be transcribed. Steroid receptor coactivator (SRC) proteins and p160 proteins with histone acetyltransferase (HAT) activity can attach to the ligand binding domain of either RAR or RXR in the presence of a ligand. The SRC coactivators acetylate the lysine residues within the core histones near the DNA binding site, neutralizing the histone charge and weakening the interaction with the chromatin. The chromatin relaxes and becomes more accessible for the DNA transcription mechanism.9
ATP-dependent chromatin remodeling proteins such as SWI/SNF use the energy of ATP hydrolysis to reposition nucleosome binding at the gene promoter region, forming nucleosome-free or nucleosome-spaced regions and providing access for the transcription machinery. An example of additional coactivating or cointegrating proteins is the p300/CRB protein. p300/CRB functions within the retinoid pathway by recruiting additional general transcription factors to the promoter region and by associating with transcription machinery at the initiation site of transcription (see Figure 5).7,8
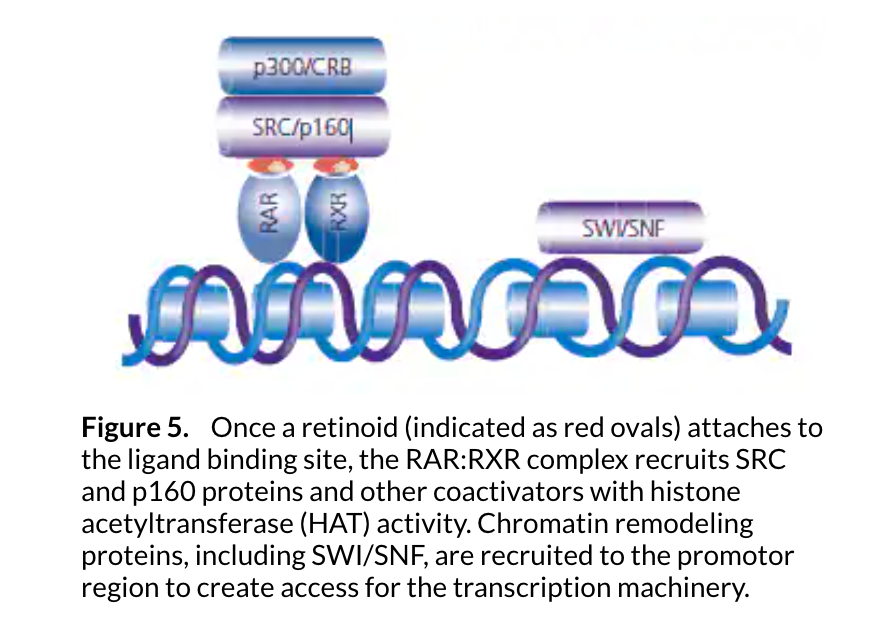
After chromatin remodeling, the coactivators dissociate, possibly to be degraded by the proteosome. The SRB and mediator containing complex (SMCC) is subsequently recruited by the retinoid receptors. The mediator complex facilitates entry of the transcriptional machinery, which includes RNA polymerase II (Pol II), the key enzyme in gene transcription, to the start site of the gene promoter. Chromatin remodeling also facilitates the binding of additional transcription factors. RA initiates the TNF-α-induced binding of NF-κB and works synergistically with TNF-α to recruit and stimulate the phosphorylation of Pol II that has been associated with enzyme activation (see Figure 6).13
REGULATION OF GENE TRANSCRIPTION
The variety of physiological process influenced by RA may be due in part to the possible combinations and redundancies within the RAR and RXR receptor families. While RARs are the primary factors for initiation of the retinoid pathway, RXRs may contribute a synergistic activity to the process in addition to mediating RARE attachment by RARs.5 In a definitive study, Balmer and Blomhoff reviewed the published data on 532 genes regulated by RA. Their analysis of the literature included evaluating which genes had strong evidence of regulation by the classical pathway.12 Only 27 genes were strongly identified as being regulated by the classical pathway, of which 26 were upregulated and one was variably regulated. The number of genes upregulated by RA, regardless of pathway was 311; interestingly 212 genes were either downregulated or variably regulated. The direct, classical pathway, while it provides insight into the function of RA, is not the only method by which RA influences expression.
Reviews of the RA pathway often include ubiquitination and phosphorylation as downstream processes that are crucial to gene regulation and transcription.1,7,8 Degradation of RAR/RXR by the proteosome after ubiquitination terminates retinoid signaling, so ubiquitination may mediate transcription by initiating a termination process. Phosphorylation is of importance as RAR and RXR isoforms can function as kinase substrates. Phosphorylation may participate in proteosome degradation and affect the ability of the holo- RAR:RXR complex to associate with coregulators and general transcription factors.8 Phosphorylation of the serine residues of RARγ has been shown to control both the RARγ transactivation function and the degradation of RARγ by the proteosome.7
The expression of intermediate proteins may provide a mechanistic explanation of the indirect regulation of transcription. RA may regulate gene expression independently of its binding to RAREs, via regulation of other transcription factors.1 This indirect regulation could explain the downregulation of genes of interest due to regulation and expression of interfering proteins.
RIP140 is a corepressor protein that preferentially binds to ligand-attached RAR, rather than to the apo-RAR:RXR complex that typically recruits corepressor proteins. RIP140 inhibits the transactivation function of several ligand-bound nuclear receptors and recruits HDACs to RAR:RXR-bound gene promoters. RIP140 may recruit HDACs to other proteins for use as substrates or may itself function as a substrate for HDAC. This selective binding by the corepressor RIP140 has been speculated as providing a regulatory mechanism for retinoid signaling.10,14 Recent work using NT2/D1 cells treated with RIP140 siRNA found measurable changes in the expression levels of RA-dependent genes but not of RA-independent genes.15
In addition to the RAR and RXR receptor families, RA functions as a ligand for peroxisome proliferator-activated receptor PPAR β/δ, a nuclear receptor that induces cell survival genes.11 The fatty acid binding protein 5 (FABP5) acts as a chaperone to transfer RA to the PPAR β/δ receptor. RA distribution to nuclear RAR and PPAR β/δ is regulated by the transfer proteins CRABP-II and FABP5, respectively, and increased expression of FABP5 preferentially shifts the transfer of RA to PPAR β/δ.21,23 MCF-7 cells that expressed a high concentration of fatty acid-binding protein 5 (FABP5) for use as a RA chaparone relative to the CRABP-II concentration were associated with cell survival and proliferation.11 In addition to nuclear gene transcription, RA has been shown to mediate transcription of mitochondrial genes; while the mechanism has not been defined, it has been theorized to involve both direct and indirect expression pathways.16
Since RXR is able to form heterodimers with other nuclear receptors, including other PPARs, cross-talk with other signaling pathways mediated by PPAR can occur.7,8 9-cis-RA can serve as the single activating ligand for PPAR:RXR, and 9-cis-RA and other rexinoids (compounds that are specific ligands for RXR) can initiate PPAR signaling independent of PPAR ligands via activation of the PPAR:RXR dimer.7,8 In addition, non-provitamin A carotenoids may interact with PPAR receptors, since PPARs can be activated by a variety of lipophilic molecules.17 Retinoic acid have been shown to target the P13K/Akt pathway, with early increased activity of Akt but a later-induced decrease in Akt activity.18 Other alternative mechanisms of retinoic acid influence are likely still to be discovered.
The endogenous level of retinoic acid is crucial for normal development because of the large number of genes affected by RA signaling. Extreme concentration levels are teratogenic to the differentiating cell, and a prescribed concentration range is necessary for normal embryonic development. Synthetic retinoids, rexinoids, and retinoic acid metabolism blocking agents (RAMBAs) are currently being investigated for their potential value as RAR/RXR ligands in modifying signals, blocking the metabolism of RA by cytochrome P450 enyzmes (CYPs), and raising endogenous levels of RA.19,20
References
1.Blomhoff R, Blomhoff HK. 2006. Overview of retinoid metabolism and function. J. J.. 66(7):606-630. http://dx.doi.org/10.1002/neu.20242
2.McCaffery PJ, Adams J, Maden M, Rosa-Molinar E. 2003. Too much of a good thing: retinoic acid as an endogenous regulator of neural differentiation and exogenous teratogen. Eur J Neurosci. 18(3):457-472. http://dx.doi.org/10.1046/j.1460-9568.2003.02765.x
3.Wyss A. 2004. Carotene Oxygenases: A New Family of Double Bond Cleavage Enzymes. 134(1):246S-250S. http://dx.doi.org/10.1093/jn/134.1.246s
4.Lakshman MR. 2004. Alpha and Omega of Carotenoid Cleavage. 134(1):241S-245S. http://dx.doi.org/10.1093/jn/134.1.241s
5.Chambon P. 2005. The Nuclear Receptor Superfamily: A Personal Retrospect on the First Two Decades. 19(6):1418-1428. http://dx.doi.org/10.1210/me.2005-0125
6.Krust A, Kastner P, Petkovich M, Zelent A, Chambon P. 1989. A third human retinoic acid receptor, hRAR-gamma.. Proceedings of the National Academy of Sciences. 86(14):5310-5314. http://dx.doi.org/10.1073/pnas.86.14.5310
7.Bastien J, Rochette-Egly C. 2004. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene. 3281-16. http://dx.doi.org/10.1016/j.gene.2003.12.005
8.McGrane MM. 2007. Vitamin A regulation of gene expression: molecular mechanism of a prototype gene. The Journal of Nutritional Biochemistry. 18(8):497-508. http://dx.doi.org/10.1016/j.jnutbio.2006.10.006
9.KISHIMOTO M, FUJIKI R, TAKEZAWA S, SASAKI Y, NAKAMURA T, YAMAOKA K, KITAGAWA H, KATO S. 2006. Nuclear Receptor Mediated Gene Regulation through Chromatin Remodeling and Histone Modifications. Endocr J. 53(2):157-172. http://dx.doi.org/10.1507/endocrj.53.157
10.Wei L. 2004. Retinoids and Receptor Interacting Protein 140 (RIP140) in Gene Regulation. CMC. 11(12):1527-1532. http://dx.doi.org/10.2174/0929867043365017
11.Schug TT, Berry DC, Shaw NS, Travis SN, Noy N. 2007. Opposing Effects of Retinoic Acid on Cell Growth Result from Alternate Activation of Two Different Nuclear Receptors. Cell. 129(4):723-733. http://dx.doi.org/10.1016/j.cell.2007.02.050
12.Balmer JE, Blomhoff R. 2002. Gene expression regulation by retinoic acid. J. Lipid Res.. 43(11):1773-1808. http://dx.doi.org/10.1194/jlr.r100015-jlr200
13.Witcher M, Pettersson F, Dupere-Richer D, Padovani A, Summers-Deluca L, Baldwin AS, Miller WH. Retinoic acid modulates chromatin to potentiate tumor necrosis factor alpha signaling on the DIF2 promoter. Nucleic Acids Research. 36(2):435-443. http://dx.doi.org/10.1093/nar/gkm1058
14.White KA, Yore MM, Warburton SL, Vaseva AV, Rieder E, Freemantle SJ, Spinella MJ. 2003. Negative Feedback at the Level of Nuclear Receptor Coregulation. J. Biol. Chem.. 278(45):43889-43892. http://dx.doi.org/10.1074/jbc.c300374200
15.Heim KC, White KA, Deng D, Tomlinson CR, Moore JH, Freemantle SJ, Spinella MJ. 2007. Selective repression of retinoic acid target genes by RIP140 during induced tumor cell differentiation of pluripotent human embryonal carcinoma cells. Mol Cancer. 6(1):57. http://dx.doi.org/10.1186/1476-4598-6-57
16.Berdanier CD. 2006. Mitochondrial Gene Expression: Influence of Nutrients and Hormones. Exp Biol Med (Maywood). 231(10):1593-1601. http://dx.doi.org/10.1177/153537020623101003
17.Bertram JS, Vine AL. 2005. Cancer prevention by retinoids and carotenoids: Independent action on a common target. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1740(2):170-178. http://dx.doi.org/10.1016/j.bbadis.2005.01.003
18.Bastien J, Plassat J, Payrastre B, Rochette-Egly C. 2006. The phosphoinositide 3-kinase/Akt pathway is essential for the retinoic acid-induced differentiation of F9 cells. Oncogene. 25(14):2040-2047. http://dx.doi.org/10.1038/sj.onc.1209241
19.Lippman SM, Lotan R. 2000. Advances in the Development of Retinoids as Chemopreventive Agents. 130(2):479S-482S. http://dx.doi.org/10.1093/jn/130.2.479s
20.Njar VC, Gediya L, Purushottamachar P, Chopra P, Vasaitis TS, Khandelwal A, Mehta J, Huynh C, Belosay A, Patel J. 2006. Retinoic acid metabolism blocking agents (RAMBAs) for treatment of cancer and dermatological diseases. Bioorganic & Medicinal Chemistry. 14(13):4323-4340. http://dx.doi.org/10.1016/j.bmc.2006.02.041
21.Wolf G. Retinoic acid as cause of cell proliferation or cell growth inhibition depending on activation of one of two different nuclear receptors. 66(1):55-59. http://dx.doi.org/10.1111/j.1753-4887.2007.00006.x
22.Perissi V, Rosenfeld MG. 2005. Controlling nuclear receptors: the circular logic of cofactor cycles. Nat Rev Mol Cell Biol. 6(7):542-554. http://dx.doi.org/10.1038/nrm1680
23.Schug TT, Berry DC, Toshkov IA, Cheng L, Nikitin AY, Noy N. 2008. Overcoming retinoic acid-resistance of mammary carcinomas by diverting retinoic acid from PPAR / to RAR. Proceedings of the National Academy of Sciences. 105(21):7546-7551. http://dx.doi.org/10.1073/pnas.0709981105