






{kind=link}
{kind=link}
Determine the necessary mass, volume, or concentration for preparing a solution.
Basic Description
| Storage Temp | Store at -20°C,Avoid repeated freezing and thawing | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Shipped In | Ice chest + Ice pads | ||||||||||||||||||||||||||||
| Product Description | Products content
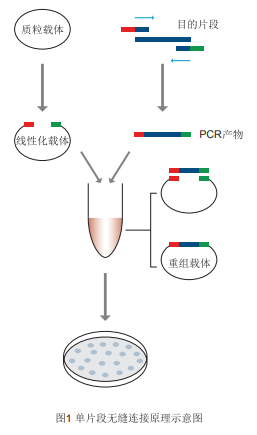
Product Description Seamless Cloning Technology is a simple, efficient and fast DNA cloning technology that allows targeted cloning of DNA insert fragments into any site of any vector. This product does not depend on T4 ligase, and is not restricted by the truncation and enzymatic sites of the target fragment, but directly uses the homologous recombination method, using a special enzyme to recombine the vector after linearization by any method with the PCR fragment with 20-25 bp overlapping region at both ends of the linearized vector, and the directional and seamless cloning of 1-5 fragments can be quickly realized in 5-15 minutes by 50C reaction.
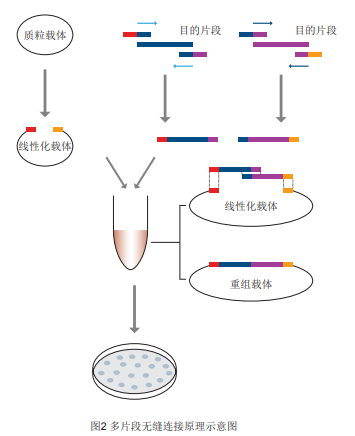
Product Features 1.15 minutes to insert one or more long or short PCR amplicons (flat/A-end) into the vector. 2. Cloning is not limited by the availability of vector and insert cleavage sites and flat/sticky ends, and can be performed at any site. 3. Seamless cloning, the insertion point does not introduce unwanted base sequences. 4. High efficiency and accuracy, clone positive rate >95%. Prepare your own materials and reagents -Insertion fragments, specific primers, linearized vectors -High-fidelity PCR reagents (Super Pfx MasterMix recommended) -Receptor Cells (DH5α Competent Cell, TOP10 Competent Cell) -Gel Recovery Kit (Rapid Agarose Gel DNA Recovery Kit) -PCR instrument, PCR reaction tubes, etc. Preparation of linearized vectors and insert DNA fragments for recovery 1. Preparation of linearized carriers for recycling (1) Select a suitable cloning site to linearize the vector. There are two ways to linearize the vector: restriction endonuclease digestion and reverse PCR amplification. The linear vector obtained by digestion, single or double digestion is acceptable, after digestion, it is recommended to use PCR product purification kit or gel recovery kit to purify the vector. Since there is no DNA ligase in the Seamless Cloning System, it will not cause the vector to self-associate. False-positive clones (without insert fragments) after recombinant product transformation are formed by incomplete cleavage of non-linearized cyclic vectors. Therefore, we recommend purification by gel recovery after cleavage to minimize the proportion of non-linearized vectors. (2) When the linearized vector is obtained by reverse PCR amplification, it is recommended to use high-fidelity polymerase amplification to reduce the introduction of amplification mutations. When using cyclic plasmid as template, it is recommended to use the endonuclease Dpn I to digest the PCR product to reduce the cloning false positives caused by the residual cyclic plasmid template. If Dpn I is used to digest the plasmid template, it is necessary to inactivate the activity of Dpn I by heating at 80℃ for 20 minutes to avoid the degradation of the cloning vector by residual Dpn I during the recombination reaction. 2. Preparation and recovery of insert fragments The preparation of insert fragments can be amplified by any PCR enzyme, and the cloning process is not affected by the flat or sticky ends (A-tails) of the amplified product (which will be removed during recombination and will not be present in the final cloned product), but in order to minimize the number of amplification mutations, especially in point mutation experiments, it is recommended to use a high fidelity polymerase (Super Pfx Master Mix) for amplification. In general, it is recommended that the PCR product be purified by gel recovery to reduce the proportion of background. If the insert fragment is derived from a plasmid template and the plasmid has the same resistance as the recombinant vector, the PCR product should be digested with the endonuclease Dpn I to reduce the background and increase the positive rate. 3. Principles of primer design for insertion fragments Principle of single fragment primer design: introduce homologous sequences at the 5' end of the primer for forward and reverse amplification of the inserted fragment, so that the amplified inserted fragment carries homologous sequences corresponding to the two ends of the linearized vector (20-25 bp without enzyme cleavage site). 5'-upstream vector terminal homologous sequence (20 bp) + digestion site (can be deleted) + gene-specific forward amplification primer Sequence (20 bp)-3' 5'-downstream vector terminal homologous sequence (20 bp) + digestion site (can be deleted) + gene-specific reverse amplification primer Sequence (20 bp)-3' Principle of multi-fragment primer design: The primer design principle at both ends of the vector is the same as that of single fragment cloning, and the primer for overlapping region between fragments is designed. Principle of multi-fragment primer design: the reverse primer of Fragment A contains a 20-25 bp overlap region with the forward primer of Fragment B and a specific primer region, the reverse primer of Fragment B contains a 20-25 bp overlap region with the forward primer of Fragment C and a specific primer region, and so on, and the primers of the two ends of the fragments contain homologous sequences of the two ends of the linear vectors. As shown:
procedure
1. Amount of linearized vectors and insert fragments used 1.1 Vectors are generally 20-50 ng, with an optimal molar ratio of insert fragments to vector between 2:1 and 3:1; in the case of multiple fragment ligation .1, the fragment-to-fragment molar ratio is 1:1. 1.2 Single fragment cloning usage Optimal cloning vector usage = (0.02 x number of cloning vector base pairs) ng (0.03 pmol) Optimal insertion fragment usage = (0.04 x number of insertion fragment base pairs) ng (0.06 pmol) 1.3 Multi-fragment cloning usage (2-5 fragments) Optimal cloning vector usage = (0.02 x number of cloning vector base pairs) ng (0.03 pmol) Usage per insert fragment = (0.02 x number of base pairs per insert fragment) ng (0.03 pmol) 2. Reorganization reactions Note: The 2×Cloning MasterMix contains PEG, which is very viscous, and even more so when it is taken out of the refrigerator at low temperatures. You can reduce the viscosity by quickly thawing it in the palm of your hand (this does not affect the quality), and then mixing it with a gentle flick. 2.1 Set up the reaction system according to the following table (can be prepared at room temperature using EP tubes)
2.2 Control reaction system (optional) 2.3. Mix gently and react at 50°C in a water bath or PCR instrument for 5-15 minutes, or for ligations of more than 3 fragments, the reaction time can be extended 2.3. to 30 minutes. At the end of the reaction, cool the EP tubes on ice and transform them directly or store them at 2.3. -20℃. 3. Reorganization product transformation 3.1 Grams of the sensory state was thawed on ice, and 10 uI of the reaction product was added to 100 uI of the sensory cell, and the wall of the tube was flicked. Mix well and let stand on ice for 30 minutes. 3.2 Heat-stimulate in a 42 cc water bath for 90 seconds and immediately place on ice for 2-3 minutes. 3.3 Add 600 ul of LB liquid medium without antimicrobial resistance and incubate at 220 rpm for 30 min at 37C. 3.4 Centrifuge at 5000 rpm for 5 min, discard excess culture medium, resuspend the organisms with the remaining 100 ul of culture medium, and use a sterile blotting rod. Spread evenly on the correctly resistant plate. 3.537'CC incubator inverted incubation for 12-16 hours 4. Identification of positive clones Depending on the specific situation, we can choose colony PCR monitoring, plasmid extraction for restriction endonuclease monitoring or sequencing identification. Bring your own instruments Thermostatic mixer. Pre-experiment Preparation and Important Notes 1. Read these instructions carefully before experimenting. 2. If Proteinase K is to be stored for a long period of time, please keep it at -20℃. 3. Check Buffer RLC for crystallization or precipitation prior to use, and if crystallization or precipitation occurs, redissolve Buffer RLC in a 56°C water bath. 4. Pre-treatment of tissue samples: Take 20 mg of tissue samples into 1.5 mL centrifuge tubes (self-provided), add 500 μL of Buffer RLC, and after the tissue homogenizer breaks up, centrifuge the samples for 1 minute at 12,000 rpm (~13,400×g), and take 200 μL of supernatant as samples.
procedure 1. Take a 1.5 mL centrifuge tube (provided), add 500 μL of Buffer RLC, 200 μL of sample, 20 μL of Proteinase K, vortex for 5 s, and then place it in a thermostatic mixer at 1200 rpm for 10 min at room temperature. Note: For wet swab samples, 200 μL of sample was taken after sufficiently shaking and mixing. Note: For wet swab samples, 200 μL was taken from the sample after it was soaked in 400 μL of saline, shaken and mixed thoroughly for 5 minutes, and then centrifuged at 12,000 rpm for 1 minute, and 200 μL was taken for extraction. 2. Instantly remove the centrifuge tube and add the solution from step 1 to the Spin Columns DM in the collection tube. centrifuge at 12,000 rpm (~13,400 x g) for 1 minute, pour off the waste liquid from the collection tube, and return the column to the collection tube. 3. Add 500 μL of Buffer PGWT to the adsorbent column, centrifuge at 12,000 rpm for 1 minute, pour off the waste liquid from the collection tube, and return the column to the collection tube. 4. Add 500 μL of Buffer GWT2 to the adsorbent column, centrifuge at 12,000 rpm for 1 minute, pour off the waste liquid from the collection tube, and return the column to the collection tube. 5. Centrifuge at 12,000 rpm for 2 minutes and pour off the waste liquid in the collection tube. Place the adsorption column at room temperature for 2 minutes and allow to dry. 6. Place the column in a new collection tube (RNase-Free Centrifuge Tube), add 40-100 μL of RNase-Free Water to the center of the column membrane, let it stand at room temperature for 2 minutes, and then centrifuge at 12,000 rpm for 1 minute to collect the nucleic acid solution. Store at -80℃ for a long time.
|
Certificates
Certificate of Analysis(COA)
Enter Lot Number to search for COA: